- Record: found
- Abstract: found
- Article: found
Morphological characteristics and phylogenetic evidence reveal two new species of Acremonium (Hypocreales, Sordariomycetes)
Abstract
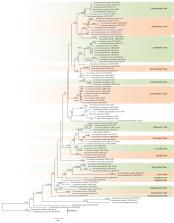
Using chicken feathers as bait, Acremonium globosisporum sp. nov. and Acremonium curvum sp. nov. were collected from the soil of Yuncheng East Garden Wildlife Zoo and Zhengzhou Zoo in China. They were identified by combining the morphological characteristics and the two-locus DNA sequence (LSU and ITS) analyses. In the phylogenetic tree, both new species clustered into separate subclades, respectively. They were different from their allied species in their morphology. The description, illustrations, and phylogenetic tree of the two new species were provided.
Related collections
Most cited references28

- Record: found
- Abstract: found
- Article: found
MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability

- Record: found
- Abstract: found
- Article: found
IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies
- Record: found
- Abstract: found
- Article: found