- Record: found
- Abstract: found
- Article: found
The quest towards understanding the molecular pathogenesis of triplet repeat disorders: Huntingtons Disease and Fragile X-Associated Tremor and Ataxia Syndrome
Author Summary
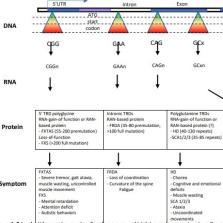
Summary
Trinucleotide repeat disorders (TRDs) make up a group of neurological inherited diseases mediated by unstable protein expansions. Polyglutamine expansion lead to diseases such as Huntington’s Disease (HD), while polyglycine expansions lead to diseases such as Fragile-X Associated Tremor and Ataxia Syndrome (FXTAS). The normal huntingtin gene contains 6-34 CAG repeats; however, upon a threshold effect that exceeds 36 repeats, HD becomes fully penetrant and promotes degeneration of neuronal populations. In contrast, FXTAS arises from polyglycine repeats between 22-200. FXTAS carriers experience severe tremor, ataxia and brain atrophy. Here, we analyze the various ways mutant huntingtin and FMR1 mRNA aggregates induce intracellular dysfunction in HD and FXTAS, specifically in the context of impaired neuronal processes and protein-protein interactions. The identification of these molecular processes offers potential targets for drug intervention that can lead to the development of innovative therapies to treat HD and FXTAS neuropathogenesis.