- Record: found
- Abstract: found
- Article: found
FunGeneNet: a web tool to estimate enrichment of functional interactions in experimental gene sets
Read this article at
Abstract
Background
Estimation of functional connectivity in gene sets derived from genome-wide or other biological experiments is one of the essential tasks of bioinformatics. A promising approach for solving this problem is to compare gene networks built using experimental gene sets with random networks. One of the resources that make such an analysis possible is CrossTalkZ, which uses the FunCoup database. However, existing methods, including CrossTalkZ, do not take into account individual types of interactions, such as protein/protein interactions, expression regulation, transport regulation, catalytic reactions, etc., but rather work with generalized types characterizing the existence of any connection between network members.
Results
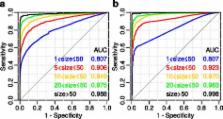
We developed the online tool FunGeneNet, which utilizes the ANDSystem and STRING to reconstruct gene networks using experimental gene sets and to estimate their difference from random networks. To compare the reconstructed networks with random ones, the node permutation algorithm implemented in CrossTalkZ was taken as a basis. To study the FunGeneNet applicability, the functional connectivity analysis of networks constructed for gene sets involved in the Gene Ontology biological processes was conducted. We showed that the method sensitivity exceeds 0.8 at a specificity of 0.95. We found that the significance level of the difference between gene networks of biological processes and random networks is determined by the type of connections considered between objects. At the same time, the highest reliability is achieved for the generalized form of connections that takes into account all the individual types of connections. By taking examples of the thyroid cancer networks and the apoptosis network, it is demonstrated that key participants in these processes are involved in the interactions of those types by which these networks differ from random ones.
Conclusions
FunGeneNet is a web tool aimed at proving the functionality of networks in a wide range of sizes of experimental gene sets, both for different global networks and for different types of interactions. Using examples of thyroid cancer and apoptosis networks, we have shown that the links over-represented in the analyzed network in comparison with the random ones make possible a biological interpretation of the original gene/protein sets. The FunGeneNet web tool for assessment of the functional enrichment of networks is available at http://www-bionet.sscc.ru/fungenenet/.
Related collections
Most cited references50
- Record: found
- Abstract: found
- Article: not found
A comprehensive two-hybrid analysis to explore the yeast protein interactome.
- Record: found
- Abstract: found
- Article: not found
Integrative approaches for finding modular structure in biological networks.
- Record: found
- Abstract: found
- Article: not found