- Record: found
- Abstract: found
- Article: found
UNC13A in amyotrophic lateral sclerosis: from genetic association to therapeutic target
Read this article at
Abstract
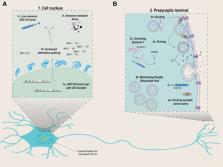
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease with limited treatment options and an incompletely understood pathophysiology. Although genomewide association studies (GWAS) have advanced our understanding of the disease, the precise manner in which risk polymorphisms contribute to disease pathogenesis remains unclear. Of relevance, GWAS have shown that a polymorphism (rs12608932) in the UNC13A gene is associated with risk for both ALS and frontotemporal dementia (FTD). Homozygosity for the C-allele at rs12608932 modifies the ALS phenotype, as these patients are more likely to have bulbar-onset disease, cognitive impairment and FTD at baseline as well as shorter survival. UNC13A is expressed in neuronal tissue and is involved in maintaining synaptic active zones, by enabling the priming and docking of synaptic vesicles. In the absence of functional TDP-43, risk variants in UNC13A lead to the inclusion of a cryptic exon in UNC13A messenger RNA, subsequently leading to nonsense mediated decay, with loss of functional protein. Depletion of UNC13A leads to impaired neurotransmission. Recent discoveries have identified UNC13A as a potential target for therapy development in ALS, with a confirmatory trial with lithium carbonate in UNC13A cases now underway and future approaches with antisense oligonucleotides currently under consideration. Considering UNC13A is a potent phenotypic modifier, it may also impact clinical trial outcomes. This present review describes the path from the initial discovery of UNC13A as a risk gene in ALS to the current therapeutic options being explored and how knowledge of its distinct phenotype needs to be taken into account in future trials.
Related collections
Most cited references101

- Record: found
- Abstract: found
- Article: found
The mutational constraint spectrum quantified from variation in 141,456 humans
- Record: found
- Abstract: found
- Article: not found
THE GENETICS OF CAENORHABDITIS ELEGANS
- Record: found
- Abstract: found
- Article: not found