- Record: found
- Abstract: found
- Article: found
A comparative genomic analysis of putative pathogenicity genes in the host-specific sibling species Colletotrichum graminicola and Colletotrichum sublineola
Read this article at
Abstract
Background
Colletotrichum graminicola and C. sublineola cause anthracnose leaf and stalk diseases of maize and sorghum, respectively. In spite of their close evolutionary relationship, the two species are completely host-specific. Host specificity is often attributed to pathogen virulence factors, including specialized secondary metabolites (SSM), and small-secreted protein (SSP) effectors. Genes relevant to these categories were manually annotated in two co-occurring, contemporaneous strains of C. graminicola and C. sublineola. A comparative genomic and phylogenetic analysis was performed to address the evolutionary relationships among these and other divergent gene families in the two strains.
Results
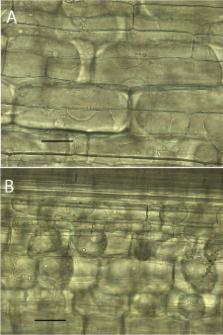
Inoculation of maize with C. sublineola, or of sorghum with C. graminicola, resulted in rapid plant cell death at, or just after, the point of penetration. The two fungal genomes were very similar. More than 50% of the assemblies could be directly aligned, and more than 80% of the gene models were syntenous. More than 90% of the predicted proteins had orthologs in both species. Genes lacking orthologs in the other species (non-conserved genes) included many predicted to encode SSM-associated proteins and SSPs. Other common groups of non-conserved proteins included transporters, transcription factors, and CAZymes. Only 32 SSP genes appeared to be specific to C. graminicola, and 21 to C. sublineola. None of the SSM-associated genes were lineage-specific. Two different strains of C. graminicola, and three strains of C. sublineola, differed in no more than 1% percent of gene sequences from one another.
Conclusions
Efficient non-host recognition of C. sublineola by maize, and of C. graminicola by sorghum, was observed in epidermal cells as a rapid deployment of visible resistance responses and plant cell death. Numerous non-conserved SSP and SSM-associated predicted proteins that could play a role in this non-host recognition were identified. Additional categories of genes that were also highly divergent suggested an important role for co-evolutionary adaptation to specific host environmental factors, in addition to aspects of initial recognition, in host specificity. This work provides a foundation for future functional studies aimed at clarifying the roles of these proteins, and the possibility of manipulating them to improve management of these two economically important diseases.
Related collections
Most cited references106
- Record: found
- Abstract: found
- Article: not found
The Sorghum bicolor genome and the diversification of grasses.
- Record: found
- Abstract: found
- Article: not found
Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2).
- Record: found
- Abstract: found
- Article: not found