- Record: found
- Abstract: found
- Article: found
Evolutionary and Molecular Analysis of Complete Genome Sequences of Norovirus From Brazil: Emerging Recombinant Strain GII.P16/GII.4
Read this article at
Abstract
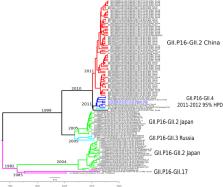
Noroviruses (NoVs) are enteric viruses that cause acute gastroenteritis, and the pandemic GII.4 genotype is spreading and evolving rapidly. The recombinant GII.P16/GII.4_Sydney strain emerged in 2016, replacing GII.P31/GII.4_Sydney (GII.P31 formerly known as GII.Pe) in some countries. We analyzed the complete genome of 20 NoV strains (17 GII.P31/GII.4_ Sydney and 3 GII.P16/GII.4_Sydney) from Belém and Manaus, Brazil, collected from 2012 to 2016. Phylogenetic trees were constructed by maximum likelihood method from 191 full NoV-VP1 sequences, demonstrated segregation of the Sydney lineage in two larger clades, suggesting that GII.4 strains associated with GII.P16 already have modifications compared with GII.P31/GII.4. Additionally, the Bayesian Markov Chain Monte Carlo method was used to reconstruct a time-scaled phylogenetic tree formed by GII.P16 ORF1 sequences ( n = 117) and three complete GII.P16 sequences from Belém. The phylogenetic tree indicated the presence of six clades classified into different capsid genotypes and locations. Evolutionary rates of the ORF1 gene of GII.P16 strains was estimated at 2.01 × 10 –3 substitutions/site/year, and the most recent common ancestors were estimated in 2011 (2011–2012, 95% HPD). Comparing the amino acid (AA) sequence coding for ORF1 with the prototype strain GII.P16/GII.4, 36 AA changes were observed, mainly in the non-structural proteins p48, p22, and RdRp. GII.P16/GII.4 strains of this study presented changes in amino acids 310, 333, 373, and 393 of the antigenic sites in the P2 subdomain, and ML tree indicating the division within the Sydney lineage according to the GII.P16 and GII.P31 polymerases. Notably, as noroviruses have high recombination rates and the GII.4 genotype was prevalent for a long time in several locations, additional and continuous evolutionary analyses of this new genotype should be needed in the future.
Related collections
Most cited references57

- Record: found
- Abstract: found
- Article: found
MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability
- Record: found
- Abstract: found
- Article: found
RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies

- Record: found
- Abstract: found
- Article: found