- Record: found
- Abstract: found
- Article: not found
A comparative study of 5- fluorouracil, doxorubicin, methotrexate, paclitaxel for their inhibition ability for Mpro of nCoV: Molecular docking and molecular dynamics simulations
Read this article at

Abstract
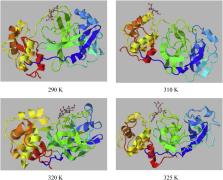
A new corona virus (nCoV) is aetiological agent responsible for the viral pneumonia epidemic. Three is no specific therapeutic medicines available for the treatment of this condition and also effective treatment choices are few. In this work author tried to investigate some repurposing drug such as 5- fluorouracil, doxorubicin, methotrexate and paclitaxel against the main protease (Mpro) of nCoV by the computational model. Molecular docking was performed to screen out the best compound and doxorubicin was found to have minimum binding energy −121.89 kcal/mol. To further study, MD simulations were performed at 300 K and the result successfully corroborate the energy obtained by molecular docking. Temperature dependent MD simulation of the best molecule that is doxorubicin obtained from docking result was performed to check the variation in structural changes in Mpro of nCoV at 290 K, 310 K, 320 K and 325 K. It is sound that doxorubicin binds effectively with Mpro of nCoV at 290 K. Further ADME properties of the 5- fluorouracil, doxorubicin, methotrexate and paclitaxel were also evaluated to understand the bioavailability.
Graphical abstract

Related collections
Most cited references65
- Record: found
- Abstract: not found
- Article: not found
GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers

- Record: found
- Abstract: found
- Article: found
SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules
- Record: found
- Abstract: found
- Article: not found