- Record: found
- Abstract: found
- Article: found
Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay
Read this article at
Abstract
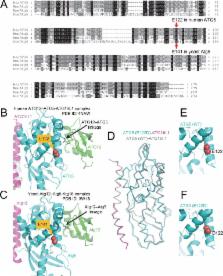
Autophagy is required for the homeostasis of cellular material and is proposed to be involved in many aspects of health. Defects in the autophagy pathway have been observed in neurodegenerative disorders; however, no genetically-inherited pathogenic mutations in any of the core autophagy-related ( ATG) genes have been reported in human patients to date. We identified a homozygous missense mutation, changing a conserved amino acid, in ATG5 in two siblings with congenital ataxia, mental retardation, and developmental delay. The subjects' cells display a decrease in autophagy flux and defects in conjugation of ATG12 to ATG5. The homologous mutation in yeast demonstrates a 30-50% reduction of induced autophagy. Flies in which Atg5 is substituted with the mutant human ATG5 exhibit severe movement disorder, in contrast to flies expressing the wild-type human protein. Our results demonstrate the critical role of autophagy in preventing neurological diseases and maintaining neuronal health.
eLife digest
Ataxia is a rare disease that affects balance and co-ordination, leading to difficulties in walking and other movements. The disease mostly affects adults, but some children are born with it and they often have additional cognitive and developmental problems. Mutations in at least 60 genes are known to be able to cause ataxia, but it is thought that there are still more to be found.
Kim, Sandford et al. studied two siblings with the childhood form of ataxia and found that they both had a mutation in a gene called ATG5. The protein produced by the mutant ATG5 gene was less able to interact with another protein called ATG12. Furthermore, the cells of both children had defects in a process called autophagy – which destroys old and faulty proteins to prevent them accumulating and causing damage to the cell.
Next, Kim, Sandford et al. examined the effect of this mutation in baker’s yeast cells. Cells with a mutation in the yeast equivalent of human ATG5 had lower levels of autophagy than normal cells. Further experiments used fruit flies that lacked fly Atg5, which were unable to fly or walk properly. Inserting the normal form of human ATG5 into the flies restored normal movement, but the mutant form of the gene had less of an effect.
These findings suggest that a mutation in ATG5 can be responsible for the symptoms of childhood ataxia. Kim, Sandford et al. think that other people with severe ataxia may have mutations in genes involved in autophagy. Therefore, the next step is to study autophagy in cells from many other ataxia patients.
Related collections
Most cited references37
- Record: found
- Abstract: found
- Article: not found
An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases.
- Record: found
- Abstract: found
- Article: not found
Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein.
- Record: found
- Abstract: found
- Article: not found