- Record: found
- Abstract: found
- Article: found
Genome-wide association study to identify genomic regions and positional candidate genes associated with male fertility in beef cattle
Read this article at
Abstract
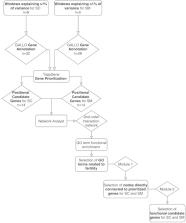
Fertility plays a key role in the success of calf production, but there is evidence that reproductive efficiency in beef cattle has decreased during the past half-century worldwide. Therefore, identifying animals with superior fertility could significantly impact cow-calf production efficiency. The objective of this research was to identify candidate regions affecting bull fertility in beef cattle and positional candidate genes annotated within these regions. A GWAS using a weighted single-step genomic BLUP approach was performed on 265 crossbred beef bulls to identify markers associated with scrotal circumference (SC) and sperm motility (SM). Eight windows containing 32 positional candidate genes and five windows containing 28 positional candidate genes explained more than 1% of the genetic variance for SC and SM, respectively. These windows were selected to perform gene annotation, QTL enrichment, and functional analyses. Functional candidate gene prioritization analysis revealed 14 prioritized candidate genes for SC of which MAP3K1 and VIP were previously found to play roles in male fertility. A different set of 14 prioritized genes were identified for SM and five were previously identified as regulators of male fertility ( SOD2, TCP1, PACRG, SPEF2, PRLR). Significant enrichment results were identified for fertility and body conformation QTLs within the candidate windows. Gene ontology enrichment analysis including biological processes, molecular functions, and cellular components revealed significant GO terms associated with male fertility. The identification of these regions contributes to a better understanding of fertility associated traits and facilitates the discovery of positional candidate genes for future investigation of causal mutations and their implications.
Related collections
Most cited references83
- Record: found
- Abstract: found
- Article: not found
PLINK: a tool set for whole-genome association and population-based linkage analyses.

- Record: found
- Abstract: found
- Article: found
ToppGene Suite for gene list enrichment analysis and candidate gene prioritization
- Record: found
- Abstract: found
- Article: not found