- Record: found
- Abstract: found
- Article: found
A subgroup of light-driven sodium pumps with an additional Schiff base counterion
Read this article at
Abstract
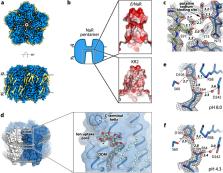
Light-driven sodium pumps (NaRs) are unique ion-transporting microbial rhodopsins. The major group of NaRs is characterized by an NDQ motif and has two aspartic acid residues in the central region essential for sodium transport. Here we identify a subgroup of the NDQ rhodopsins bearing an additional glutamic acid residue in the close vicinity to the retinal Schiff base. We thoroughly characterize a member of this subgroup, namely the protein ErNaR from Erythrobacter sp. HL-111 and show that the additional glutamic acid results in almost complete loss of pH sensitivity for sodium-pumping activity, which is in contrast to previously studied NaRs. ErNaR is capable of transporting sodium efficiently even at acidic pH levels. X-ray crystallography and single particle cryo-electron microscopy reveal that the additional glutamic acid residue mediates the connection between the other two Schiff base counterions and strongly interacts with the aspartic acid of the characteristic NDQ motif. Hence, it reduces its pKa. Our findings shed light on a subgroup of NaRs and might serve as a basis for their rational optimization for optogenetics.
Abstract
Light-driven sodium-pumping rhodopsins are unique ion transporters. Here, authors present a characterization of such rhodopsins with a modified active center allowing for efficient sodium transport under various environmental conditions.
Related collections
Most cited references68

- Record: found
- Abstract: found
- Article: found
Highly accurate protein structure prediction with AlphaFold
- Record: found
- Abstract: found
- Article: not found
UCSF Chimera--a visualization system for exploratory research and analysis.
- Record: found
- Abstract: found
- Article: not found