- Record: found
- Abstract: found
- Article: found
Uromodulin-related autosomal-dominant tubulointerstitial kidney disease—pathogenetic insights based on a case
Read this article at
Abstract
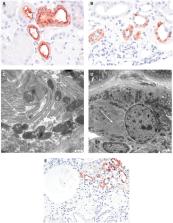
Uromodulin-related autosomal-dominant tubulointerstitial kidney disease (ADTKD-UMOD) is a rare monogenic disorder that is characterized by tubulointerstitial fibrosis and progression of kidney function loss, and may progress to end-stage renal disease. It is usually accompanied by hyperuricaemia and gout. Mutations in the uromodulin gene ( UMOD) resulting in malfunctioning of UMOD are known to be the cause of ADTKD-UMOD, which is assumed to be an endoplasmatic reticulum (ER) storage disease. As a case vignette, we report a 29-year-old female with a suspicious family history of chronic kidney disease presenting with progressive loss of renal function, hyperuricaemia and frequent urinary tract infections. Urinary tract infections and pyelonephritides may represent a clinical feature of uromodulin malfunction as it plays a protective role against urinary tract infections despite only sporadic data on this topic. ADTKD-UMOD was diagnosed after genetic testing revealing a missense mutation in the UMOD gene. Light microscopy showed excessive tubular interstitial fibrosis and tubular atrophy together with signs of glomerular sclerosis. Electron microscopic findings could identify electron dense storage deposits in the ER of tubular epithelial cells of the thick ascending loop. Immunohistological staining with KDEL (lysine, aspartic acid, glutamic acid, leucine) showed positivity in the tubular cells, which likely represents ER expansion upon accumulation of misfolded UMOD which could trigger the unfolded protein response and ER stress. This review highlights pathophysiological mechanisms that are subject to ADTKD-UMOD.
Related collections
Most cited references57
- Record: found
- Abstract: found
- Article: not found
PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress.
- Record: found
- Abstract: found
- Article: not found
Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy.
- Record: found
- Abstract: found
- Article: not found