- Record: found
- Abstract: found
- Article: not found
Computationally Driven, Quantitative Experiments Discover Genes Required for Mitochondrial Biogenesis
Read this article at

Abstract
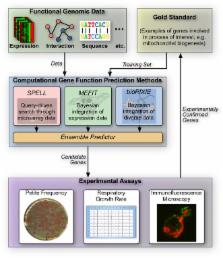
Mitochondria are central to many cellular processes including respiration, ion homeostasis, and apoptosis. Using computational predictions combined with traditional quantitative experiments, we have identified 100 proteins whose deficiency alters mitochondrial biogenesis and inheritance in Saccharomyces cerevisiae. In addition, we used computational predictions to perform targeted double-mutant analysis detecting another nine genes with synthetic defects in mitochondrial biogenesis. This represents an increase of about 25% over previously known participants. Nearly half of these newly characterized proteins are conserved in mammals, including several orthologs known to be involved in human disease. Mutations in many of these genes demonstrate statistically significant mitochondrial transmission phenotypes more subtle than could be detected by traditional genetic screens or high-throughput techniques, and 47 have not been previously localized to mitochondria. We further characterized a subset of these genes using growth profiling and dual immunofluorescence, which identified genes specifically required for aerobic respiration and an uncharacterized cytoplasmic protein required for normal mitochondrial motility. Our results demonstrate that by leveraging computational analysis to direct quantitative experimental assays, we have characterized mutants with subtle mitochondrial defects whose phenotypes were undetected by high-throughput methods.
Author Summary
Mitochondria are the proverbial powerhouses of the cell, running the fundamental biochemical processes that produce energy from nutrients using oxygen. These processes are conserved in all eukaryotes, from humans to model organisms such as baker's yeast. In humans, mitochondrial dysfunction plays a role in a variety of diseases, including diabetes, neuromuscular disorders, and aging. In order to better understand fundamental mitochondrial biology, we studied genes involved in mitochondrial biogenesis in the yeast S. cerevisiae, discovering over 100 proteins with novel roles in this process. These experiments assigned function to 5% of the genes whose function was not known. In order to achieve this rapid rate of discovery, we developed a system incorporating highly quantitative experimental assays and an integrated, iterative process of computational protein function prediction. Beginning from relatively little prior knowledge, we found that computational predictions achieved about 60% accuracy and rapidly guided our laboratory work towards hundreds of promising candidate genes. Thus, in addition to providing a more thorough understanding of mitochondrial biology, this study establishes a framework for successfully integrating computation and experimentation to drive biological discovery. A companion manuscript, published in PLoS Computational Biology (doi:10.1371/journal.pcbi.1000322), discusses observations and conclusions important for the computational community.
Related collections
Most cited references79
- Record: found
- Abstract: found
- Article: not found
Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae.
- Record: found
- Abstract: found
- Article: not found
Automatic clustering of orthologs and in-paralogs from pairwise species comparisons.
- Record: found
- Abstract: found
- Article: not found