- Record: found
- Abstract: found
- Article: found
Development of a Single Nucleotide Polymorphism Barcode to Genotype Plasmodium vivax Infections
Read this article at
Abstract
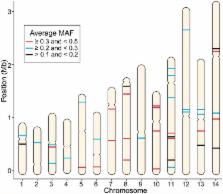
Plasmodium vivax, one of the five species of Plasmodium parasites that cause human malaria, is responsible for 25–40% of malaria cases worldwide. Malaria global elimination efforts will benefit from accurate and effective genotyping tools that will provide insight into the population genetics and diversity of this parasite. The recent sequencing of P. vivax isolates from South America, Africa, and Asia presents a new opportunity by uncovering thousands of novel single nucleotide polymorphisms (SNPs). Genotyping a selection of these SNPs provides a robust, low-cost method of identifying parasite infections through their unique genetic signature or barcode. Based on our experience in generating a SNP barcode for P. falciparum using High Resolution Melting (HRM), we have developed a similar tool for P. vivax. We selected globally polymorphic SNPs from available P. vivax genome sequence data that were located in putatively selectively neutral sites (i.e., intergenic, intronic, or 4-fold degenerate coding). From these candidate SNPs we defined a barcode consisting of 42 SNPs. We analyzed the performance of the 42-SNP barcode on 87 P. vivax clinical samples from parasite populations in South America (Brazil, French Guiana), Africa (Ethiopia) and Asia (Sri Lanka). We found that the P. vivax barcode is robust, as it requires only a small quantity of DNA (limit of detection 0.3 ng/μl) to yield reproducible genotype calls, and detects polymorphic genotypes with high sensitivity. The markers are informative across all clinical samples evaluated (average minor allele frequency > 0.1). Population genetic and statistical analyses show the barcode captures high degrees of population diversity and differentiates geographically distinct populations. Our 42-SNP barcode provides a robust, informative, and standardized genetic marker set that accurately identifies a genomic signature for P. vivax infections.
Author Summary
Plasmodium vivax malaria is a major global public health problem, with nearly 2.5 billion people at risk for infection and approximately 132–391 million clinical infections annually. It has a wide geographical range, with a high disease burden in Asia, Central and South America, the Middle East, Oceania, and East Africa. Advances in sequencing technology and sample processing have made it possible to characterize the genetic diversity of P. vivax populations. This genetic variation provides a means to identify parasites by unique genetic signatures, or “barcodes.” We developed such a genetic barcode for P. vivax, composed of 42 robust and informative variants. Here we report its development and validation based on 87 clinical samples identified by microscopy to contain P. vivax from geographically diverse parasite populations from South America (Brazil, French Guiana), Africa (Ethiopia) and Asia (Sri Lanka). We show that the SNP barcode provides a genotyping tool that can be performed at low cost, providing a means to uniquely identify parasite infections and distinguish geographic origins, and that barcode data may offer new insights into P. vivax population structure and diversity.
Related collections
Most cited references36
- Record: found
- Abstract: found
- Article: not found
Comparative genomics of the neglected human malaria parasite Plasmodium vivax.
- Record: found
- Abstract: found
- Article: not found
Vivax malaria: neglected and not benign.
- Record: found
- Abstract: found
- Article: not found