- Record: found
- Abstract: found
- Article: found
P53 is Subjected to Lipoteichoic Acid-Induced Phosphorylation in the Lungs
letter
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
P53 is a transcription factor protecting the cells against malignancies via modulation
of multifarious regulatory signaling cascades. Those activities may result to either
cellular repair, or to the elimination of the irreversible damaged tissue components.
Recent evidence suggest that this endothelium defender (P53) exerts strong anti-inflammatory
activities in the lungs.
1
P53 protects the endothelium cells against the lipopolysaccharide (LPS)-induced endothelial
hyperpermeability by reducing the generation of the reactive oxygen species,
2
by suppressing the inflammatory RhoA/MLC2 pathway,
3
and by inducing the repairing activities of the unfolded protein response in the lungs.
4
5
Lung endothelial barrier dysfunction is both a cause and a consequence of severe lung
inflammatory disease, including the lethal acute respiratory distress syndrome (ARDS).
1
Indeed, P53 expression levels are crucial for the integrity of the lung microvasculature,
since P53 reduction due to LPS-induced P53 phosphorylation or small interfering ribonucleic
acid has been previously shown to be related to the collapse of the lung barrier function.
6
Lipoteichoic acid (LTA) contributes in ARDS.
7
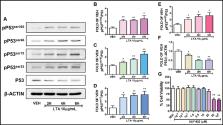
Fig. 1A
demonstrates by Western blotting in bovine pulmonary arterial endothelial cells purchased
from Genlantis (PB30205) (San Diego, California, United States) that LTA induces the
phosphorylation of P53 and suppresses its expression levels. The LTA from
Staphylococcus aureus
(L2515) was purchased from Sigma-Aldrich (St. Louis, Missouri, United States). The
densitometric analysis performed with Image J software indicated that this toxin,
which is a major constituent of the cell wall of Gram-positive bacteria, increases
the expression of pP53
ser392
(
Fig. 1B
), pP53
ser46
(
Fig. 1C
), pP53
ser15
(
Fig. 1D
), and pP53
ser33
(
Fig. 1E
), and reduces P53 (
Fig. 1F
). Interestingly, Hsp90 inhibitors are anticancer agents, which have been shown to
counteract the LPS-induced P53 degradation, and deliver protective effects in the
inflamed lungs.
3
Although those compounds were initially developed to stochastically eliminate cancers,
it now appears (
Fig. 1G
) that they do not affect the viability of human lung microvascular cells (HuLEC-5a)
(CRL-3244), which were obtained from the American Type Culture Collection (Manassas,
Virginia, United States). Details regarding cell cultures and Western blotting have
been previously reported.
2
4
Fig. 1
(
A
–
F
) Western blot analysis of phosphorylated P53 (pP53
ser392
, pP53
ser46
, pP53
ser15
, pP53
ser33
) and total P53 expression after treatment of bovine pulmonary artery endothelial
cell (BPAEC) with either lipoteichoic acid (LTA) (10 µg/mL) or vehicle (VEH) (phosphate-buffered
saline [PBS]) for 2, 4, and 6 hours. The blots shown are representative of three independent
experiments. The signal intensity of the protein bands was analyzed by densitometry.
Protein levels of phosphorylated P53 and P53 were normalized to P53 and β-actin, respectively.
*
p
< 0.05, **
p
< 0.01 vs. VEH. Means ± standard error of mean (SEM). (
G
) Effects of the Hsp90 inhibitor AUY-922 in the viability of HuLEC-5a. Cells were
treated with either VEH (0.1% dimethyl sulfoxide [DMSO]) or AUY-922 (10
−3
, 10
−2
, 10
−1
, 1.0, 5.0, 10.0, 25.0, 50.0, 100 µM) for 24 hours. Cell viability was evaluated by
employing the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
**
p
< 0.01 vs. VEH,
n
= 3. Means ± SEM.
Treatment of HuLEC-5a cells with moderate concentrations of AUY-922 (101756–820) (
Fig. 1G
) from VWR (Radnor, Pennsylvania, United States) did not affect the viability of those
cells, as measured with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT) assay. Briefly, the cells were seeded in 96-well culture plates (10,000 cells/well)
in complete growth media and were treated with AUY-922 (0–100 µM). After 24 hours,
the media was replaced with fresh media containing 0.5 mg/mL MTT. After 3.5 hours
of incubation, dimethyl sulfoxide (100 μL/well) was added to dissolve the MTT crystals,
and 15 minutes later the absorbance was measured at 570 nm in a Synergy H1 Hybrid
Multi-Mode Reader from Biotek (Winooski, Vermont, United States). In all cases, GraphPad
Prism (version 5.01) was used to analyze the data, and the values are expressed as
the mean ± standard error of mean. Values of
p
less than 0.05 were considered as an indication of statistical significance, and the
number of experimental repeats is indicated by the letter
n
.
In conclusion, the present letter aims to substantiate our hypothesis that P53 is
a target of the “inflammatory storm”-induced ARDS. Thus, pharmacological induction
of P53 due to treatments with Hsp90 inhibitors, or growth hormone releasing hormone
antagonists
8
may deliver a promising approach against the severe lung inflammatory disease.
9
Related collections
Most cited references7
- Record: found
- Abstract: found
- Article: not found
Lung inflammation induced by lipoteichoic acid or lipopolysaccharide in humans.
Tom van der Poll, Christian Draing, Femke de Vrij … (2008)
- Record: found
- Abstract: not found
- Article: not found
Hsp90 inhibitors induce the unfolded protein response in bovine and mice lung cells
- Record: found
- Abstract: found
- Article: not found