- Record: found
- Abstract: found
- Article: found
Single-cell RNA-seq of Drosophila miranda testis reveals the evolution and trajectory of germline sex chromosome regulation
Read this article at
Abstract
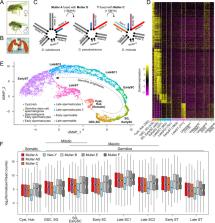
Although sex chromosomes have evolved from autosomes, they often have unusual regulatory regimes that are sex- and cell-type-specific such as dosage compensation (DC) and meiotic sex chromosome inactivation (MSCI). The molecular mechanisms and evolutionary forces driving these unique transcriptional programs are critical for genome evolution but have been, in the case of MSCI in Drosophila, subject to continuous debate. Here, we take advantage of the younger sex chromosomes in D. miranda (XR and the neo-X) to infer how former autosomes acquire sex-chromosome-specific regulatory programs using single-cell and bulk RNA sequencing and ribosome profiling, in a comparative evolutionary context. We show that contrary to mammals and worms, the X down-regulation through germline progression is most consistent with the shutdown of DC instead of MSCI, resulting in half gene dosage at the end of meiosis for all 3 X’s. Moreover, lowly expressed germline and meiotic genes on the neo-X are ancestrally lowly expressed, instead of acquired suppression after sex linkage. For the young neo-X, DC is incomplete across all tissue and cell types and this dosage imbalance is rescued by contributions from Y-linked gametologs which produce transcripts that are translated to compensate both gene and protein dosage. We find an excess of previously autosomal testis genes becoming Y-specific, showing that the neo-Y and its masculinization likely resolve sexual antagonism. Multicopy neo-sex genes are predominantly expressed during meiotic stages of spermatogenesis, consistent with their amplification being driven to interfere with mendelian segregation. Altogether, this study reveals germline regulation of evolving sex chromosomes and elucidates the consequences these unique regulatory mechanisms have on the evolution of sex chromosome architecture.
Abstract
Although sex chromosomes have evolved from autosomes, they often have unusual regulatory regimes that are sex- and cell-type-specific. This study takes advantage of the young sex chromosomes of Drosophila miranda to explore how former autosomes acquire sex chromosome-specific regulatory programs, revealing the consequences that these mechanisms have for the evolution of sex chromosome architecture.
Related collections
Most cited references101
- Record: found
- Abstract: found
- Article: not found
STAR: ultrafast universal RNA-seq aligner.

- Record: found
- Abstract: found
- Article: found
Fast and accurate short read alignment with Burrows–Wheeler transform

- Record: found
- Abstract: found
- Article: found