- Record: found
- Abstract: found
- Article: found
MetaboAnalyst 6.0: towards a unified platform for metabolomics data processing, analysis and interpretation

Read this article at
Abstract
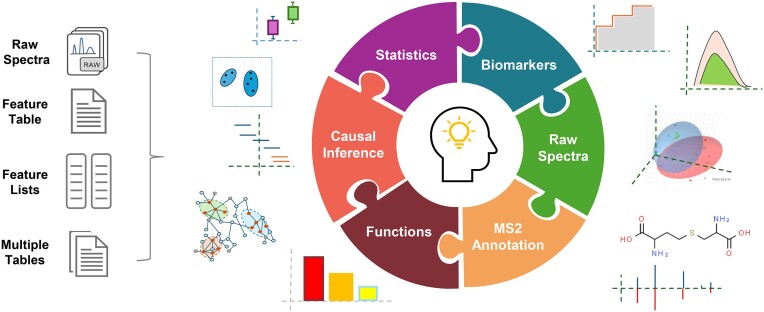
We introduce MetaboAnalyst version 6.0 as a unified platform for processing, analyzing, and interpreting data from targeted as well as untargeted metabolomics studies using liquid chromatography - mass spectrometry (LC–MS). The two main objectives in developing version 6.0 are to support tandem MS (MS2) data processing and annotation, as well as to support the analysis of data from exposomics studies and related experiments. Key features of MetaboAnalyst 6.0 include: (i) a significantly enhanced Spectra Processing module with support for MS2 data and the asari algorithm; (ii) a MS2 Peak Annotation module based on comprehensive MS2 reference databases with fragment-level annotation; (iii) a new Statistical Analysis module dedicated for handling complex study design with multiple factors or phenotypic descriptors; (iv) a Causal Analysis module for estimating metabolite - phenotype causal relations based on two-sample Mendelian randomization, and (v) a Dose-Response Analysis module for benchmark dose calculations. In addition, we have also improved MetaboAnalyst's visualization functions, updated its compound database and metabolite sets, and significantly expanded its pathway analysis support to around 130 species. MetaboAnalyst 6.0 is freely available at https://www.metaboanalyst.ca.
Graphical Abstract
Related collections
Most cited references74

- Record: found
- Abstract: found
- Article: found
limma powers differential expression analyses for RNA-sequencing and microarray studies

- Record: found
- Abstract: found
- Article: found
The MR-Base platform supports systematic causal inference across the human phenome

- Record: found
- Abstract: found
- Article: found