- Record: found
- Abstract: found
- Article: found
duper is a null mutation of Cryptochrome 1 in Syrian hamsters
Read this article at
Significance
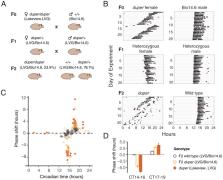
We successfully identified the duper allele as a null mutation of Cryptochrome 1 in Syrian hamsters. Here, we have shown the use of fast homozygosity mapping as an effective approach to identify causal mutations in mammals, despite lacking chromosomal genome information. In the course of this work, we improved the draft Syrian hamster genome and generated datasets necessary to exploit Syrian hamsters as a modern genetic research model. The unique physiological features of Syrian hamsters make them a desirable model to investigate human diseases, including circadian disorders, cancer, heart function, metabolism, and infectious diseases (e.g., severe acute respiratory syndrome coronavirus 2).
Abstract
The duper mutation is a recessive mutation that shortens the period length of the circadian rhythm in Syrian hamsters. These animals show a large phase shift when responding to light pulses. Limited genetic resources for the Syrian hamster ( Mesocricetus auratus) presented a major obstacle to cloning duper. This caused the duper mutation to remain unknown for over a decade. In this study, we did a de novo genome assembly of Syrian hamsters with long-read sequencing data from two different platforms, Pacific Biosciences and Oxford Nanopore Technologies. Using two distinct ecotypes and a fast homozygosity mapping strategy, we identified duper as an early nonsense allele of Cryptochrome 1 ( Cry1) leading to a short, unstable protein. CRY1 is known as a highly conserved component of the repressive limb of the core circadian clock. The genome assembly and other genomic datasets generated in this study will facilitate the use of the Syrian hamster in biomedical research.
Related collections
Most cited references54

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools

- Record: found
- Abstract: found
- Article: found
Fast and accurate short read alignment with Burrows–Wheeler transform
- Record: found
- Abstract: found
- Article: not found
The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data.
Author and article information
Comments
Comment on this article
See how this article has been cited at scite.ai
scite shows how a scientific paper has been cited by providing the context of the citation, a classification describing whether it supports, mentions, or contrasts the cited claim, and a label indicating in which section the citation was made.