- Record: found
- Abstract: found
- Article: found
Gene Deregulation and Underlying Mechanisms in Spinocerebellar Ataxias With Polyglutamine Expansion
Read this article at
Abstract
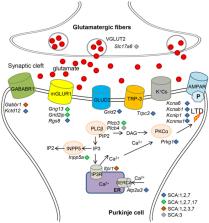
Polyglutamine spinocerebellar ataxias (polyQ SCAs) include SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17 and constitute a group of adult onset neurodegenerative disorders caused by the expansion of a CAG repeat sequence located within the coding region of specific genes, which translates into polyglutamine tract in the corresponding proteins. PolyQ SCAs are characterized by degeneration of the cerebellum and its associated structures and lead to progressive ataxia and other diverse symptoms. In recent years, gene and epigenetic deregulations have been shown to play a critical role in the pathogenesis of polyQ SCAs. Here, we provide an overview of the functions of wild type and pathogenic polyQ SCA proteins in gene regulation, describe the extent and nature of gene expression changes and their pathological consequences in diseases, and discuss potential avenues to further investigate converging and distinct disease pathways and to develop therapeutic strategies.
Related collections
Most cited references196
- Record: found
- Abstract: found
- Article: not found
Epigenetic Mechanisms of Longevity and Aging.
- Record: found
- Abstract: found
- Article: not found
Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1.
- Record: found
- Abstract: found
- Article: found