- Record: found
- Abstract: found
- Article: found
Expression of SRP-9001 dystrophin and stabilization of motor function up to 2 years post-treatment with delandistrogene moxeparvovec gene therapy in individuals with Duchenne muscular dystrophy

Read this article at
Abstract
Introduction: Delandistrogene moxeparvovec (SRP-9001) is an investigational gene transfer therapy designed for targeted expression of SRP-9001 dystrophin protein, a shortened dystrophin retaining key functional domains of the wild-type protein.
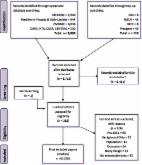
Methods: This Phase 2, double-blind, two-part (48 weeks per part) crossover study (SRP-9001-102 [Study 102]; NCT03769116) evaluated delandistrogene moxeparvovec in patients, aged ≥4 to <8 years with Duchenne muscular dystrophy. Primary endpoints (Part 1) were change from baseline (CFBL) in SRP-9001 dystrophin expression (Week 12), by Western blot, and in North Star Ambulatory Assessment (NSAA) score (Week 48). Safety assessments included treatment-related adverse events (TRAEs). Patients were randomized and stratified by age to placebo (n = 21) or delandistrogene moxeparvovec (n = 20) and crossed over for Part 2.
Results: SRP-9001 dystrophin expression was achieved in all patients: mean CFBL to Week 12 was 23.82% and 39.64% normal in Parts 1 and 2, respectively. In Part 1, CFBL to Week 48 in NSAA score (least-squares mean, LSM [standard error]) was +1.7 (0.6) with treatment versus +0.9 (0.6) for placebo; p = 0.37. Disparity in baseline motor function between groups likely confounded these results. In 4- to 5-year-olds with matched baseline motor function, CFBL to Week 48 in NSAA scores was significantly different (+2.5 points; p = 0.0172), but not significantly different in 6-to-7-year-olds with imbalanced baseline motor function (−0.7 points; p = 0.5384). For patients treated with delandistrogene moxeparvovec in Part 2, CFBL to Week 48 in NSAA score was +1.3 (2.7), whereas for those treated in Part 1, NSAA scores were maintained. As all patients in Part 2 were exposed to treatment, results were compared with a propensity-score-weighted external control (EC) cohort. The LSM difference in NSAA score between the Part 2 treated group and EC cohort was statistically significant (+2.0 points; p = 0.0009). The most common TRAEs were vomiting, decreased appetite, and nausea. Most occurred within the first 90 days and all resolved.
Discussion: Results indicate robust expression of SRP-9001 dystrophin and overall stabilization in NSAA up to 2 years post-treatment. Differences in NSAA between groups in Part 1 were not significant for the overall population, likely because cohorts were stratified only by age, and other critical prognostic factors were not well matched at baseline.
Related collections
Most cited references26
- Record: found
- Abstract: found
- Article: not found
Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management
- Record: found
- Abstract: found
- Article: found
The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review

- Record: found
- Abstract: found
- Article: found