- Record: found
- Abstract: found
- Article: found
α-Solanine induces ROS-mediated autophagy through activation of endoplasmic reticulum stress and inhibition of Akt/mTOR pathway
Read this article at
Abstract
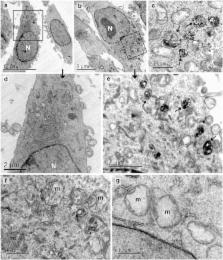
α-Solanine is a glycoalkaloid found in species of the nightshade family including potato. It was primarily reported to have toxic effects in humans. However, there is a growing body of literature demonstrating in vitro and in vivo anticancer activity of α-solanine. Most of these studies have shown activation of apoptosis as the underlying mechanism in antitumor activity of α-solanine. In this study, we report α-solanine as a potential inducer of autophagy, which may act synergistically or in parallel with apoptosis to exert its cytotoxic effect. Induction of autophagy was demonstrated by several assays including electron microscopy, immunoblotting of autophagy markers and immunofluorescence for LC3 (microtubule-associated protein 1 (MAP1) light chain-3) puncta. α-Solanine-induced autophagic flux was demonstrated by additionally enhanced – turnover of LC3-II and – accumulation of LC3-specific puncta after co-incubation of cells with either of the autophagolysosome inhibitors – chloroquine and – bafilomycin A1. We also demonstrated α-solanine-induced oxidative damage in regulating autophagy where pre-incubation of cells with reactive oxygen species (ROS) scavenger resulted in suppression of CM-H 2DCFDA (5 (and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester) fluorescence as well as decrease in LC3-II turnover. α-Solanine treatment caused an increase in the expression of endoplasmic reticulum (ER) stress proteins (BiP, activating transcription factor 6 (ATF6), X-box-binding protein 1, PERK, inositol-requiring transmembrane kinase/endonuclease 1, ATF4 and CCAAT-enhancer-binding protein (C/EBP)-homologous protein) suggesting activation of unfolded protein response pathway. Moreover, we found downregulation of phosphorylated Akt (Thr 308 and Ser 473), mammalian target of rapamycin (mTOR; Ser 2448 and Ser 2481) and 4E-BP1 (Thr 37/46) by α-solanine implying suppression of the Akt/mTOR pathway. Collectively, our results signify that α-solanine induces autophagy to exert anti-proliferative activity by triggering ER stress and inhibiting Akt/mTOR signaling pathway.
Related collections
Most cited references36
- Record: found
- Abstract: found
- Article: not found
Endoplasmic reticulum stress: cell life and death decisions.
- Record: found
- Abstract: found
- Article: not found
Autophagy is activated for cell survival after endoplasmic reticulum stress.
- Record: found
- Abstract: found
- Article: not found
Lysosomes and autophagy in cell death control.
Author and article information
Comments
Comment on this article
See how this article has been cited at scite.ai
scite shows how a scientific paper has been cited by providing the context of the citation, a classification describing whether it supports, mentions, or contrasts the cited claim, and a label indicating in which section the citation was made.