- Record: found
- Abstract: found
- Article: found
QTL-seq identifies cooked grain elongation QTLs near soluble starch synthase and starch branching enzymes in rice ( Oryza sativa L.)
Read this article at
Abstract
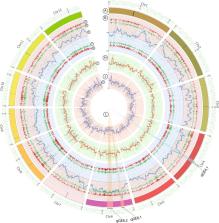
Grain quality is one of the main targets that rice breeders focus on to improve elite rice varieties. Several characteristics are considered when determine rice grain quality, such as aroma, amylose content (AC), gelatinization temperature (GT) and, especially, lengthwise grain elongation (GE). GE is a desirable feature in premium rice of high quality, such as India and Pakistan’ Basmati. Inheritance of GE in rice has not been clearly elucidated due to its complex and inconsistent pattern. In this study, we identified QTLs for GE in rice using bulk-segregant analysis (BSA) and whole-genome sequencing based on an F 2 population segregated for GE as well as AC and GT. We identified two QTLs on chromosome 6, qGE6.1 and qGE6.2, and another QTL on chromosome 4, qGE4.1. qGE6.1 and qGE6.2 were located near starch synthase IIa ( SSIIa) and starch branching enzyme III (SBEIII), respectively, and qGE4.1 was located near starch branching enzyme IIa ( SBEIIa). qGE6.1 was considered to be the major QTL for GE based on this population, and SSIIa was suggested to be the best candidate gene associated with the GE trait. The results of this study may be useful for breeding rice with increased grain elongation and different starch properties.
Related collections
Most cited references23
- Record: found
- Abstract: found
- Article: not found
QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations.
- Record: found
- Abstract: found
- Article: not found
QTL-seq identifies an early flowering QTL located near Flowering Locus T in cucumber.

- Record: found
- Abstract: found
- Article: found