- Record: found
- Abstract: found
- Article: found
Molecular Phylodynamics of the Heterosexual HIV Epidemic in the United Kingdom
Read this article at
Abstract
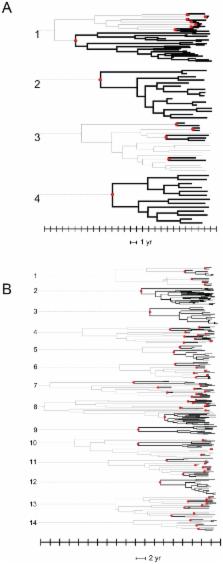
The heterosexual risk group has become the largest HIV infected group in the United Kingdom during the last 10 years, but little is known of the network structure and dynamics of viral transmission in this group. The overwhelming majority of UK heterosexual infections are of non-B HIV subtypes, indicating viruses originating among immigrants from sub-Saharan Africa. The high rate of HIV evolution, combined with the availability of a very high density sample of viral sequences from routine clinical care has allowed the phylodynamics of the epidemic to be investigated for the first time. Sequences of the viral protease and partial reverse transcriptase coding regions from 11,071 patients infected with HIV of non-B subtypes were studied. Of these, 2774 were closely linked to at least one other sequence by nucleotide distance. Including the closest sequences from the global HIV database identified 296 individuals that were in UK-based groups of 3 or more individuals. There were a total of 8 UK-based clusters of 10 or more, comprising 143/2774 (5%) individuals, much lower than the figure of 25% obtained earlier for men who have sex with men (MSM). Sample dates were incorporated into relaxed clock phylogenetic analyses to estimate the dates of internal nodes. From the resulting time-resolved phylogenies, the internode lengths, used as estimates of maximum transmission intervals, had a median of 27 months overall, over twice as long as obtained for MSM (14 months), with only 2% of transmissions occurring in the first 6 months after infection. This phylodynamic analysis of non-B subtype HIV sequences representing over 40% of the estimated UK HIV-infected heterosexual population has revealed heterosexual HIV transmission in the UK is clustered, but on average in smaller groups and is transmitted with slower dynamics than among MSM. More effective intervention to restrict the epidemic may therefore be feasible, given effective diagnosis programmes.
Author Summary
Since 1995, HIV among heterosexuals in the UK increased to the point where the total number of heterosexuals infected with HIV, predominantly of non-B subtypes, exceeds the number of HIV-positive homosexual men. To understand the dynamics of this epidemic, we have applied the novel technique of phylodynamics to the analysis of viral sequences taken in the course of routine clinical care from approximately 40% of the HIV-infected heterosexual population in the UK. Phylodynamics reconstructs the pattern of viral sequence divergence in time, revealing the size of transmission clusters and the dynamics of transmission within them. Of 11,071 patients studied, 296 were linked to at least two others in the UK. There were 8 clusters comprising 10 or more individuals among these, yielding a total of 143 or 5% of all individuals with links, much lower than seen earlier among homosexual men (25%). Viral transmissions within clusters also occurred less rapidly, only 2% being dated to the first 6 months of infection, compared to 25% among homosexual men. Overall, transmission clusters exist in the UK heterosexual HIV epidemic but they are generally smaller than among homosexuals; onward transmission occurs less rapidly and is not associated with acute HIV infection.
Related collections
Most cited references17
- Record: found
- Abstract: found
- Article: not found
MRBAYES: Bayesian inference of phylogenetic trees.
- Record: found
- Abstract: found
- Article: not found
Dating of the human-ape splitting by a molecular clock of mitochondrial DNA.
- Record: found
- Abstract: found
- Article: not found