- Record: found
- Abstract: found
- Article: found
Case report and literature review: Fabry disease misdiagnosing as polymyalgia rheumatica
Read this article at
Abstract
Rationale:
The clinical manifestations of Fabry disease affect the nerves, kidneys, heart, skin, gastrointestinal tract and eyes. Our aim is to familiarize people with the FD diagnostic process by reporting this case.
Patient concerns:
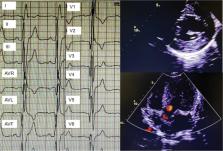
A 79-year-old-male patient presented with muscle pain and weakness in the extremities, also with an increasing erythrocyte sedimentation rate and C-reactive protein. Further examinations revealed that multiple organ involvement, such as rash, myocardial hypertrophy, peripheral neuropathy.
Diagnoses:
Cardiac MR demonstrated hypertrophic cardiomyopathy, myocardial fibrosis and low myocardial T1 value. The patient was eventually diagnosed with Fabry disease through proteomics and genetic testing.
Interventions:
The treatment is enzyme replacement therapy (ERT). But this patient could not afford ERT and was given only general symptomatic treatment, pregabalin, and a gradual reduction in glucocorticoid.
Related collections
Most cited references19
- Record: found
- Abstract: found
- Article: not found
Fabry disease revisited: Management and treatment recommendations for adult patients.
- Record: found
- Abstract: found
- Article: not found
Fabry's disease.
- Record: found
- Abstract: found
- Article: found