- Record: found
- Abstract: found
- Article: found
Next generation sequencing is a highly reliable method to analyze exon 7 deletion of survival motor neuron 1 ( SMN1) gene
Read this article at
Abstract
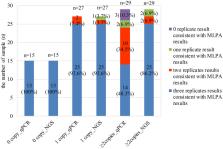
Spinal muscular atrophy (SMA) is one of the most common and severe genetic diseases. SMA carrier screening is an effective way to identify couples at risk of having affected children. Next-generation sequencing (NGS)-based expanded carrier screening could detect SMN1 gene copy number without extra experiment and with high cost performance. However, its performance has not been fully evaluated. Here we conducted a systematic comparative study to evaluate the performance of three common methods. 478 samples were analyzed with multiplex ligation probe amplification (MLPA), real-time quantitative polymerase chain reaction (qPCR) and NGS, simultaneously. Taking MLPA-based results as the reference, for 0 copy, 1 copy and ≥ 2 copy SMN1 analysis with NGS, the sensitivity, specificity and precision were all 100%. Using qPCR method, the sensitivity was 100%, 97.52% and 94.30%, respectively; 98.63%, 95.48% and 100% for specificity; and 72.72%, 88.72% and 100% for precision. NGS repeatability was higher than that of qPCR. Moreover, among three methods, NGS had the lowest retest rate. Thus, NGS is a relatively more reliable method for SMN1 gene copy number detection. In expanded carrier screening, compared with the combination of multiple methods, NGS method could reduce the test cost and simplify the screening process.
Related collections
Most cited references54
- Record: found
- Abstract: not found
- Article: not found
Identification and characterization of a spinal muscular atrophy-determining gene
- Record: found
- Abstract: found
- Article: not found
Spinal muscular atrophy.
- Record: found
- Abstract: found
- Article: not found