- Record: found
- Abstract: found
- Article: found
In-silico analysis of nonsynonymous genomic variants within CCM2 gene reaffirm the existence of dual cores within typical PTB domain
Read this article at
Abstract
Purpose
The objective of this study is to validate the existence of dual cores within the typical phosphotyrosine binding (PTB) domain and to identify potentially damaging and pathogenic nonsynonymous coding single nuclear polymorphisms (nsSNPs) in the canonical PTB domain of the CCM2 gene that causes cerebral cavernous malformations (CCMs).
Methods
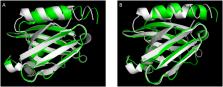
The nsSNPs within the coding sequence for PTB domain of human CCM2 gene, retrieved from exclusive database searches, were analyzed for their functional and structural impact using a series of bioinformatic tools. The effects of mutations on the tertiary structure of the PTB domain in human CCM2 protein were predicted to examine the effect of nsSNPs on the tertiary structure of PTB Cores.
Results
Our mutation analysis, through alignment of protein structures between wildtype CCM2 and mutant, predicted that the structural impacts of pathogenic nsSNPs is biophysically limited to only the spatially adjacent substituted amino acid site with minimal structural influence on the adjacent core of the PTB domain, suggesting both cores are independently functional and essential for proper CCM2 PTB function.
Conclusion
Utilizing a combination of protein conservation and structure-based analysis, we analyzed the structural effects of inherited pathogenic mutations within the CCM2 PTB domain. Our results predicted that the pathogenic amino acid substitutions lead to only subtle changes locally, confined to the surrounding tertiary structure of the PTB core within which it resides, while no structural disturbance to the neighboring PTB core was observed, reaffirming the presence of independently functional dual cores in the CCM2 typical PTB domain.
Highlights
-
•
The pathogenic amino acid mutants lead to subtle structural changes in the PTB core.
-
•
No structural disturbance to the neighboring PTB core was observed.
-
•
Data reaffirm the presence of dual functional cores in the CCM2 PTB domain.
-
•
More new genetic variants leading to CCM pathogenesis were suggested.
Related collections
Most cited references47
- Record: found
- Abstract: found
- Article: not found
UCSF Chimera--a visualization system for exploratory research and analysis.
- Record: found
- Abstract: found
- Article: not found
A method and server for predicting damaging missense mutations
- Record: found
- Abstract: not found
- Article: not found