- Record: found
- Abstract: found
- Article: found
Analysis of copy number variations in the sheep genome using 50K SNP BeadChip array
Read this article at
Abstract
Background
In recent years, genome-wide association studies have successfully uncovered single-nucleotide polymorphisms (SNPs) associated with complex traits such as diseases and quantitative phenotypes. These variations account for a small proportion of heritability. With the development of high throughput techniques, abundant submicroscopic structural variations have been found in organisms, of which the main variations are copy number variations (CNVs). Therefore, CNVs are increasingly recognized as an important and abundant source of genetic variation and phenotypic diversity.
Results
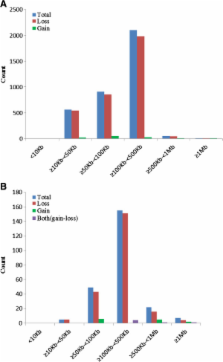
Analyses of CNVs in the genomes of three sheep breeds were performed using the Ovine SNP50 BeadChip array. A total of 238 CNV regions (CNVRs) were identified, including 219 losses, 13 gains, and six with both events (losses and gains), which cover 60.35 Mb of the sheep genomic sequence and correspond to 2.27% of the autosomal genome sequence. The length of the CNVRs on autosomes range from 13.66 kb to 1.30 Mb with a mean size of 253.57 kb, and 75 CNVRs events had a frequency > 3%. Among these CNVRs, 47 CNVRs identified by the PennCNV overlapped with the CNVpartition. Functional analysis indicated that most genes in the CNVRs were significantly enriched for involvement in the environmental response. Furthermore, 10 CNVRs were selected for validation and 6 CNVRs were further experimentally confirmed by qPCR. In addition, there were 57 CNVRs overlapped in our new dataset and other published ruminant CNV studies.
Conclusions
In this study, we firstly constructed a sheep CNV map based on the Ovine SNP50 array. Our results demonstrated the differences of two detection tools and integration of multiple algorithms can enhance the detection of sheep genomic structure variations. Furthermore, our findings would be of help for understanding the sheep genome and provide preliminary foundation for carrying out the CNVs association studies with economically important phenotypes of sheep in the future.
Related collections
Most cited references48
- Record: found
- Abstract: found
- Article: not found
Global variation in copy number in the human genome.
- Record: found
- Abstract: found
- Article: not found
Structural variation in the human genome.
- Record: found
- Abstract: found
- Article: not found