- Record: found
- Abstract: found
- Article: found
O-08 AGGRESSIVE PARAGANGLIOMA-PHEOCHROMOCYTOMA: AN UNDER-TOLD STORY OF THE SDH-D MUTATION

Read this article at
Abstract
Introduction
Hereditary paraganglioma-pheochromocytoma (PGL/PCC) syndromes are defined by the occurrence of paragangliomas derived from the autonomic nervous system and pheochromocytomas that are localized to the adrenal medulla. While pheochromocytomas and sympathetic paragangliomas cause catecholamine hypersecretion, parasympathetic paragangliomas are mostly nonsecretory. A diagnosis of hereditary PGL/PCC is based on a personal and family history of recurrent, multiple, early-onset PGL/PCC and the identification of mutations in the disease-related genes. Germline mutations in succinate dehydrogenase (SDH) genes are among the most common susceptibility genes. Some of these tumors, particularly those related to SDH-B, exhibit a significant malignant potential. However, mutations in the SDH-D gene do not often predispose to aggressive disease.
Clinical Case
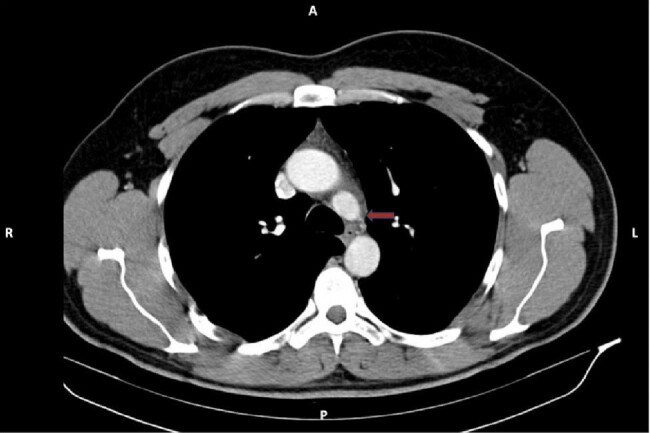
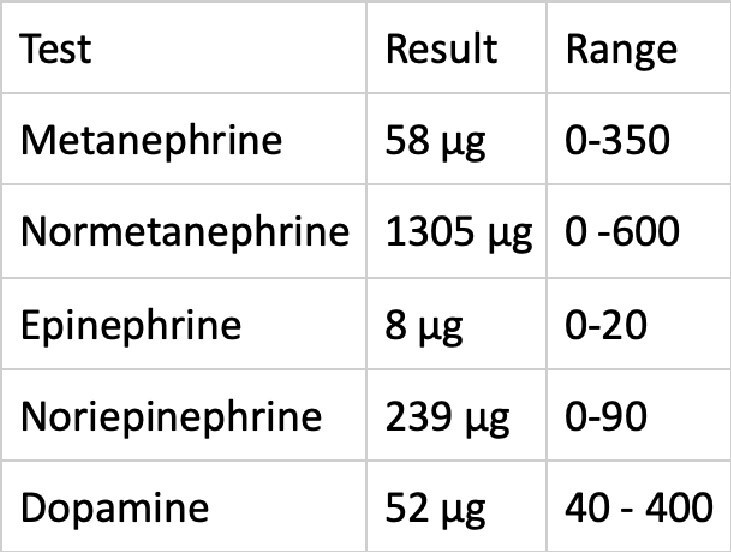
A 24-year-old male patient presented to the outpatient clinic with a headache and right flank pain. There was not family history of the disease, except for the sudden death of his father at the age of 43. An MRI of the abdomen revealed a 45x44 mm lesion in the right adrenal gland. The patient's urinary normetanephrine levels were found to be significantly elevated. Subsequently, a right adrenalectomy was performed, and the pathology report confirmed a diagnosis of pheochromocytoma. The patient was referred to our clinic after 123 - Iodine MIBG SPECT/CT scan revealed the presence of a 24x15 mm lesion at the left cervical level 3 and a 21x35 mm lesion at the right paraaortic level. Concurrent plasma and urinary catecholamine levels were within the normal range. Both were surgically excised, and the pathology result was reported as paraganglioma. The molecular analysis revealed a pathogenic SDH-D gene variant:c.147dupA (p.His50fs).The patient was started to be monitored annually. In the third year of follow-up, a 14x12 mm nodular lesion was detected in the aorticopulmonary space. The lesion was not excised due to its proximity to vital structures and the patient was continued to be monitored. During the four-year period, there was not significant growth was observed in the lesion. However, in the fifth year detection of the lesion, it's size was reaching to 28x26 mm. It was excised via thoracic surgery. The pathology result was consistent with paraganglioma, with positive surgical margins and a Ki-67 index of 6%.Despite the recommendation for genetic screening, the patient's family members declined. Ten years after the initial diagnosis, the patient's cousin presented with cervical paraganglioma at the age of 19.
Conclusion
The presented case demonstrates the significance of molecular testing for patients with PGL/PCC and the necessity of long-term follow-up. Furthermore, it suggests that individuals carrying the SDH-D gene mutation may be at an increased risk of an aggressive disease course. Therefore, the precision of genetic counseling is of vital importance in establishing clinical screening and follow-up strategies.
Related collections
Author and article information
Comments
Comment on this article
See how this article has been cited at scite.ai
scite shows how a scientific paper has been cited by providing the context of the citation, a classification describing whether it supports, mentions, or contrasts the cited claim, and a label indicating in which section the citation was made.