- Record: found
- Abstract: found
- Article: found
The Beta Amyloid Dysfunction (BAD) Hypothesis for Alzheimer’s Disease
Read this article at
Abstract
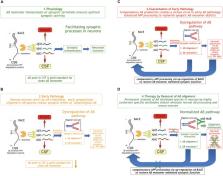
Beta amyloid, Aβ 1–42, originally named as Amyloid A4 protein, is one of the most investigated peptides in neuroscience and has attracted substantial interest since its discovery as the main insoluble fibril-type protein in cerebrovascular amyloid angiopathy ( Glenner and Wong, 1984; Masters et al., 1985) of Alzheimer’s disease (AD). From the very beginning, Aβ was regarded per se as a “bad molecule,” triggering the so-called “beta amyloid cascade hypothesis” ( Hardy and Higgins, 1992). This hypothesis ignored any physiological function for in situ generated Aβ monomer with normal production and turnover rate ( Bateman et al., 2006). Accordingly, pan-Aβ-related therapeutic approaches were designed to eliminate or lower the three structural isoforms in parallel: (1) the pre-amyloid monomer, (2) the misfolded oligomer, and (3) the final fibril. While we already knew about poor correlations between plaques and cognitive decline quite early ( Terry et al., 1991), data for an essential benign physiological role for Aβ monomer at low concentrations were also not considered to be relevant. Here, a different Beta Amyloid hypothesis is described, the so-called “Beta Amyloid Dysfunction hypothesis,” which, in contrast to the “Beta Amyloid Cascade hypothesis,” builds on the homeostasis of essential Aβ monomer in the synaptic vesicle cycle (SVC). Disease-relevant early pathology emerges through disturbance of the Aβ homeostasis by so far unknown factors leading to the formation of misfolded Aβ oligomers. These early species interfere with the synaptic physiological Aβ monomer regulation and exert their neurotoxicity via various receptors for sticky oligomer-type Aβ aggregates. The Beta Amyloid Dysfunction (BAD) hypothesis is introduced and shown to explain negative clinical results of Gamma-secretase and Beta-secretase (BACE) inhibitors as well as pan-Aβ isotype unselective immunotherapies. This hypothesis gives guidance to what needs to be done therapeutically to revive successful clinical testing in AD for this highly validated target. The BAD hypothesis will need further refinement in particular through more detailed exploration for the role of physiological Aβ monomer.
Related collections
Most cited references40
- Record: found
- Abstract: found
- Article: not found
APP processing and synaptic function.
- Record: found
- Abstract: found
- Article: not found
Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity.
- Record: found
- Abstract: found
- Article: not found