- Record: found
- Abstract: found
- Article: found
Genetic variation in adaptability and pleiotropy in budding yeast
Read this article at
Abstract
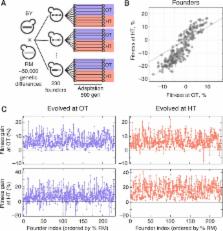
Evolution can favor organisms that are more adaptable, provided that genetic variation in adaptability exists. Here, we quantify this variation among 230 offspring of a cross between diverged yeast strains. We measure the adaptability of each offspring genotype, defined as its average rate of adaptation in a specific environmental condition, and analyze the heritability, predictability, and genetic basis of this trait. We find that initial genotype strongly affects adaptability and can alter the genetic basis of future evolution. Initial genotype also affects the pleiotropic consequences of adaptation for fitness in a different environment. This genetic variation in adaptability and pleiotropy is largely determined by initial fitness, according to a rule of declining adaptability with increasing initial fitness, but several individual QTLs also have a significant idiosyncratic role. Our results demonstrate that both adaptability and pleiotropy are complex traits, with extensive heritable differences arising from naturally occurring variation.
Related collections
Most cited references60
- Record: found
- Abstract: found
- Article: not found
The genetic landscape of a cell.
- Record: found
- Abstract: found
- Article: not found
Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq.
- Record: found
- Abstract: found
- Article: not found