- Record: found
- Abstract: found
- Article: found
Insights into the Complexity of a Dormant Mycobacterium tuberculosis Cluster Once Transmission Is Resumed
Read this article at
ABSTRACT
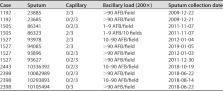
Genotyping tools help identify the complexity in Mycobacterium tuberculosis transmission clusters. We carried out a thorough analysis of the epidemiological and bacteriological complexity of a cluster in Almería, Spain. The cluster, initially associated with Moroccan migrants and with no secondary cases identified in 4 years, then reappeared in Spanish-born individuals. In one case, two Mycobacterium tuberculosis clonal variants were identified. We reanalyzed the cluster, supported by the characterization of multiple cultured isolates and respiratory specimens, whole-genome sequencing, and epidemiological case interviews. Our findings showed that the cluster, which was initially thought to have restarted activity with just a single case harboring a small degree of within-host diversity, was in fact currently growing due to coincidental reactivation of past exposures, with clonal diversity transmitted throughout the cluster. In one case, within-host diversity was amplified, probably due to prolonged diagnostic delay.
IMPORTANCE The precise study of the dynamics of tuberculosis transmission in socio-epidemiologically complex scenarios may require more thorough analysis than the standard molecular epidemiology strategies. Our study illustrates the epidemiological and bacteriological complexity present in a transmission cluster in a challenging epidemiological setting with a high proportion of migrant cases. The combination of whole-genome sequencing, refined and refocused epidemiological interviews, and in-depth analysis of the bacterial composition of sputa and cultured isolates was crucial in order to correctly reinterpret the true nature of this cluster. Our global approach allowed us to reinterpret correctly the unnoticed epidemiological and bacteriological complexity involved in the Mycobacterium tuberculosis transmission event under study, which had been overlooked by the usual molecular epidemiology approaches.
Related collections
Most cited references28
- Record: found
- Abstract: found
- Article: not found
A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3.

- Record: found
- Abstract: found
- Article: found
Fast and accurate long-read alignment with Burrows–Wheeler transform
- Record: found
- Abstract: found
- Article: not found