- Record: found
- Abstract: found
- Article: found
Efficient real-time selective genome sequencing on resource-constrained devices
Read this article at
Abstract
Background
Third-generation nanopore sequencers offer selective sequencing or “Read Until” that allows genomic reads to be analyzed in real time and abandoned halfway if not belonging to a genomic region of “interest.” This selective sequencing opens the door to important applications such as rapid and low-cost genetic tests. The latency in analyzing should be as low as possible for selective sequencing to be effective so that unnecessary reads can be rejected as early as possible. However, existing methods that employ a subsequence dynamic time warping (sDTW) algorithm for this problem are too computationally intensive that a massive workstation with dozens of CPU cores still struggles to keep up with the data rate of a mobile phone–sized MinION sequencer.
Results
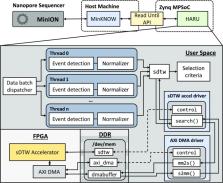
In this article, we present Hardware Accelerated Read Until (HARU), a resource-efficient hardware–software codesign-based method that exploits a low-cost and portable heterogeneous multiprocessor system-on-chip platform with on-chip field-programmable gate arrays (FPGA) to accelerate the sDTW-based Read Until algorithm. Experimental results show that HARU on a Xilinx FPGA embedded with a 4-core ARM processor is around 2.5× faster than a highly optimized multithreaded software version (around 85× faster than the existing unoptimized multithreaded software) running on a sophisticated server with a 36-core Intel Xeon processor for a SARS-CoV-2 dataset. The energy consumption of HARU is 2 orders of magnitudes lower than the same application executing on the 36-core server.
Conclusions
HARU demonstrates that nanopore selective sequencing is possible on resource-constrained devices through rigorous hardware–software optimizations. The source code for the HARU sDTW module is available as open source at https://github.com/beebdev/HARU, and an example application that uses HARU is at https://github.com/beebdev/sigfish-haru.
Related collections
Most cited references50

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools
- Record: found
- Abstract: found
- Article: not found
Minimap2: pairwise alignment for nucleotide sequences

- Record: found
- Abstract: found
- Article: found