- Record: found
- Abstract: found
- Article: found
Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis
Read this article at
Abstract
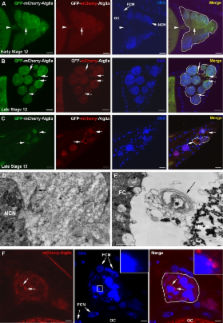
Blocking autophagy protects the apoptosis inhibitor dBruce from destruction and promotes nurse cell survival in developing egg chambers.
Abstract
Autophagy is an evolutionarily conserved pathway responsible for degradation of cytoplasmic material via the lysosome. Although autophagy has been reported to contribute to cell death, the underlying mechanisms remain largely unknown. In this study, we show that autophagy controls DNA fragmentation during late oogenesis in Drosophila melanogaster. Inhibition of autophagy by genetically removing the function of the autophagy genes atg1, atg13, and vps34 resulted in late stage egg chambers that contained persisting nurse cell nuclei without fragmented DNA and attenuation of caspase-3 cleavage. The Drosophila inhibitor of apoptosis (IAP) dBruce was found to colocalize with the autophagic marker GFP-Atg8a and accumulated in autophagy mutants. Nurse cells lacking Atg1 or Vps34 in addition to dBruce contained persisting nurse cell nuclei with fragmented DNA. This indicates that autophagic degradation of dBruce controls DNA fragmentation in nurse cells. Our results reveal autophagic degradation of an IAP as a novel mechanism of triggering cell death and thereby provide a mechanistic link between autophagy and cell death.
Related collections
Most cited references34
- Record: found
- Abstract: found
- Article: not found
Role and regulation of starvation-induced autophagy in the Drosophila fat body.
- Record: found
- Abstract: found
- Article: not found
Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain
- Record: found
- Abstract: found
- Article: not found