- Record: found
- Abstract: found
- Article: found
Flux Design: In silico design of cell factories based on correlation of pathway fluxes to desired properties
Read this article at
Abstract
Background
The identification of genetic target genes is a key step for rational engineering of production strains towards bio-based chemicals, fuels or therapeutics. This is often a difficult task, because superior production performance typically requires a combination of multiple targets, whereby the complex metabolic networks complicate straightforward identification. Recent attempts towards target prediction mainly focus on the prediction of gene deletion targets and therefore can cover only a part of genetic modifications proven valuable in metabolic engineering. Efficient in silico methods for simultaneous genome-scale identification of targets to be amplified or deleted are still lacking.
Results
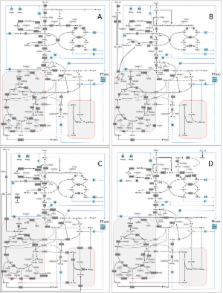
Here we propose the identification of targets via flux correlation to a chosen objective flux as approach towards improved biotechnological production strains with optimally designed fluxes. The approach, we name Flux Design, computes elementary modes and, by search through the modes, identifies targets to be amplified (positive correlation) or down-regulated (negative correlation). Supported by statistical evaluation, a target potential is attributed to the identified reactions in a quantitative manner. Based on systems-wide models of the industrial microorganisms Corynebacterium glutamicum and Aspergillus niger, up to more than 20,000 modes were obtained for each case, differing strongly in production performance and intracellular fluxes. For lysine production in C. glutamicum the identified targets nicely matched with reported successful metabolic engineering strategies. In addition, simulations revealed insights, e.g. into the flexibility of energy metabolism. For enzyme production in A.niger flux correlation analysis suggested a number of targets, including non-obvious ones. Hereby, the relevance of most targets depended on the metabolic state of the cell and also on the carbon source.
Conclusions
Objective flux correlation analysis provided a detailed insight into the metabolic networks of industrially relevant prokaryotic and eukaryotic microorganisms. It was shown that capacity, pathway usage, and relevant genetic targets for optimal production partly depend on the network structure and the metabolic state of the cell which should be considered in future metabolic engineering strategies. The presented strategy can be generally used to identify priority sorted amplification and deletion targets for metabolic engineering purposes under various conditions and thus displays a useful strategy to be incorporated into efficient strain and bioprocess optimization.
Related collections
Most cited references46
- Record: found
- Abstract: found
- Article: not found
Analysis of optimality in natural and perturbed metabolic networks.
- Record: found
- Abstract: found
- Article: not found
Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays.
- Record: found
- Abstract: found
- Article: not found