- Record: found
- Abstract: found
- Article: found
Strand-specific RNA sequencing reveals extensive regulated long antisense transcripts that are conserved across yeast species
Read this article at
Abstract
Background
Recent studies in budding yeast have shown that antisense transcription occurs at many loci. However, the functional role of antisense transcripts has been demonstrated only in a few cases and it has been suggested that most antisense transcripts may result from promiscuous bi-directional transcription in a dense genome.
Results
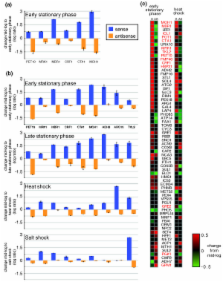
Here, we use strand-specific RNA sequencing to study anti-sense transcription in Saccharomyces cerevisiae. We detect 1,103 putative antisense transcripts expressed in mid-log phase growth, ranging from 39 short transcripts covering only the 3' UTR of sense genes to 145 long transcripts covering the entire sense open reading frame. Many of these antisense transcripts overlap sense genes that are repressed in mid-log phase and are important in stationary phase, stress response, or meiosis. We validate the differential regulation of 67 antisense transcripts and their sense targets in relevant conditions, including nutrient limitation and environmental stresses. Moreover, we show that several antisense transcripts and, in some cases, their differential expression have been conserved across five species of yeast spanning 150 million years of evolution. Divergence in the regulation of antisense transcripts to two respiratory genes coincides with the evolution of respiro-fermentation.
Related collections
Most cited references26
- Record: found
- Abstract: found
- Article: not found
The transcriptional landscape of the yeast genome defined by RNA sequencing.
- Record: found
- Abstract: found
- Article: not found
Direct multiplexed measurement of gene expression with color-coded probe pairs.
- Record: found
- Abstract: found
- Article: not found