- Record: found
- Abstract: found
- Article: found
Haplotype-resolved Chinese male genome assembly based on high-fidelity sequencing
Read this article at
Abstract
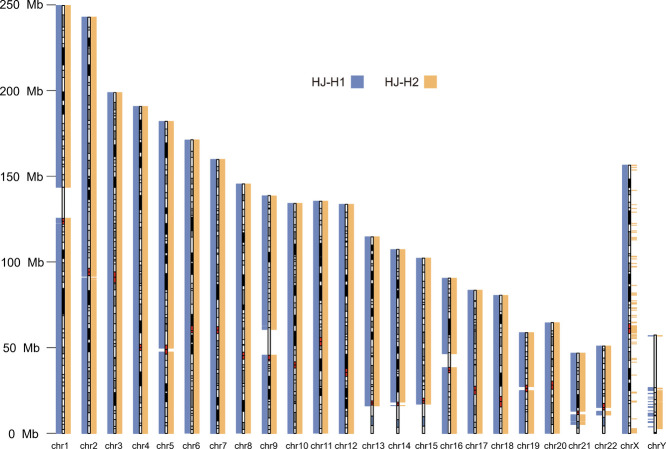
The advantages of both the length and accuracy of high-fidelity (HiFi) reads enable chromosome-scale haplotype-resolved genome assembly. In this study, we sequenced a cell line named HJ, established from a Chinese Han male individual by using HiFi and Hi-C. We assembled two high-quality haplotypes of the HJ genome (haplotype 1 (H1): 3.1 Gb, haplotype 2 (H2): 2.9 Gb). The continuity (H1: contig N50 = 28.2 Mb, H2: contig N50 = 25.9 Mb) and completeness (BUSCO: H1 = 94.9%, H2 = 93.5%) are substantially better than those of other Chinese genomes, for example, HX1, NH1.0, and YH2.0. By comparing HJ genome with GRCh38, we reported the mutation landscape of HJ and found that 176 and 213 N-gaps were filled in H1 and H2, respectively. In addition, we detected 12.9 Mb and 13.4 Mb novel sequences containing 246 and 135 protein-coding genes in H1 and H2, respectively. Our results demonstrate the advantages of HiFi reads in haplotype-resolved genome assembly and provide two high-quality haplotypes of a potential Chinese genome as a reference for the Chinese Han population.
Graphical abstract
Related collections
Most cited references49

- Record: found
- Abstract: found
- Article: found
Metascape provides a biologist-oriented resource for the analysis of systems-level datasets

- Record: found
- Abstract: found
- Article: found
A global reference for human genetic variation
- Record: found
- Abstract: found
- Article: not found