- Record: found
- Abstract: found
- Article: found
Genomic Epidemiology of Shiga Toxin-Producing Escherichia coli Isolated from the Livestock-Food-Human Interface in South America
Read this article at
Abstract
Simple Summary
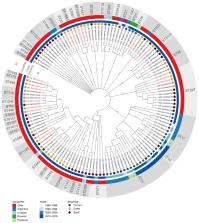
Shiga toxin-producing Escherichia coli (STEC) are zoonotic pathogens that cause food-borne diseases in humans, where cattle and derived products play a key role as reservoirs and vehicles. We analyzed the genomic data of STEC strains circulating at the livestock-food-human interface in South America, extracting clinically and epidemiologically relevant information (serotypes, virulome, resistance genes, sequence types, and phylogenomics). This study included 130 STEC genomes obtained from cattle ( n = 51), beef ( n = 48), and human ( n = 31) samples. The successful expansion of O157:H7 (ST11) and non-O157 (ST16, ST21, ST223, ST443, ST677, ST679, ST2388) clones is highlighted, suggesting common activities, such as multilateral trade and travel. Circulating STEC strains analyzed exhibit high genomic diversity and harbor several genetic determinants associated with severe illness in humans, highlighting the need to establish official surveillance of this pathogen that should be focused on detecting molecular determinants of virulence and clonal relatedness, in the whole beef production chain.
Abstract
Shiga toxin-producing Escherichia coli (STEC) are zoonotic pathogens responsible for causing food-borne diseases in humans. While South America has the highest incidence of human STEC infections, information about the genomic characteristics of the circulating strains is scarce. The aim of this study was to analyze genomic data of STEC strains isolated in South America from cattle, beef, and humans; predicting the antibiotic resistome, serotypes, sequence types (STs), clonal complexes (CCs) and phylogenomic backgrounds. A total of 130 whole genome sequences of STEC strains were analyzed, where 39.2% were isolated from cattle, 36.9% from beef, and 23.8% from humans. The ST11 was the most predicted (20.8%) and included O-:H7 (10.8%) and O157:H7 (10%) serotypes. The successful expansion of non-O157 clones such as ST16/CC29-O111:H8 and ST21/CC29-O26:H11 is highlighted, suggesting multilateral trade and travel. Virulome analyses showed that the predominant stx subtype was stx2a (54.6%); most strains carried ehaA (96.2%), iha (91.5%) and lpfA (77.7%) genes. We present genomic data that can be used to support the surveillance of STEC strains circulating at the livestock-food-human interface in South America, in order to control the spread of critical clones “from farm to table”.
Related collections
Most cited references118

- Record: found
- Abstract: found
- Article: found
Fast and accurate short read alignment with Burrows–Wheeler transform

- Record: found
- Abstract: found
- Article: found