- Record: found
- Abstract: found
- Article: found
A high-quality de novo genome assembly based on nanopore sequencing of a wild-caught coconut rhinoceros beetle ( Oryctes rhinoceros)
Read this article at
Abstract
Background
An optimal starting point for relating genome function to organismal biology is a high-quality nuclear genome assembly, and long-read sequencing is revolutionizing the production of this genomic resource in insects. Despite this, nuclear genome assemblies have been under-represented for agricultural insect pests, particularly from the order Coleoptera. Here we present a de novo genome assembly and structural annotation for the coconut rhinoceros beetle, Oryctes rhinoceros (Coleoptera: Scarabaeidae), based on Oxford Nanopore Technologies (ONT) long-read data generated from a wild-caught female, as well as the assembly process that also led to the recovery of the complete circular genome assemblies of the beetle’s mitochondrial genome and that of the biocontrol agent, Oryctes rhinoceros nudivirus (OrNV). As an invasive pest of palm trees, O. rhinoceros is undergoing an expansion in its range across the Pacific Islands, requiring new approaches to management that may include strategies facilitated by genome assembly and annotation.
Results
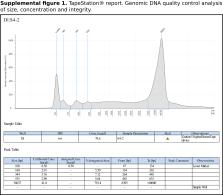
High-quality DNA isolated from an adult female was used to create four ONT libraries that were sequenced using four MinION flow cells, producing a total of 27.2 Gb of high-quality long-read sequences. We employed an iterative assembly process and polishing with one lane of high-accuracy Illumina reads, obtaining a final size of the assembly of 377.36 Mb that had high contiguity (fragment N50 length = 12 Mb) and accuracy, as evidenced by the exceptionally high completeness of the benchmarked set of conserved single-copy orthologous genes (BUSCO completeness = 99.1%). These quality metrics place our assembly ahead of the published Coleopteran genomes, including that of an insect model, the red flour beetle ( Tribolium castaneum). The structural annotation of the nuclear genome assembly contained a highly-accurate set of 16,371 protein-coding genes, with only 2.8% missing BUSCOs, and the expected number of non-coding RNAs. The number and structure of paralogous genes in a gene family like Sigma GST is lower than in another scarab beetle ( Onthophagus taurus), but higher than in the red flour beetle ( Tribolium castaneum), which suggests expansion of this GST class in Scarabaeidae. The quality of our gene models was also confirmed with the correct placement of O. rhinoceros among other members of the rhinoceros beetles (subfamily Dynastinae) in a phylogeny based on the sequences of 95 protein-coding genes in 373 beetle species from all major lineages of Coleoptera. Finally, we provide a list of 30 candidate dsRNA targets whose orthologs have been experimentally validated as highly effective targets for RNAi-based control of several beetles.
Conclusions
The genomic resources produced in this study form a foundation for further functional genetic research and management programs that may inform the control and surveillance of O. rhinoceros populations, and we demonstrate the efficacy of de novo genome assembly using long-read ONT data from a single field-caught insect.
Related collections
Most cited references79

- Record: found
- Abstract: found
- Article: found
Trimmomatic: a flexible trimmer for Illumina sequence data

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools
- Record: found
- Abstract: found
- Article: found