- Record: found
- Abstract: found
- Article: not found
Liver and Biliary System
chapter-article
M. Grant Maxie , DVM, PhD, Diplomate ACVP
5 February 2016
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Acknowledgments
The authors acknowledge the major contributions of Dr. W. Roger Kelly and Dr. M.A.
(Tony) Hayes as previous authors of this section. In addition, the authors thank Drs.
W. Roger Kelly and Jeremy Allen for critical review, discussions, and provision of
images.
General Considerations
From a contemporary perspective, the liver is a marvel of biology. It is the guardian
of homeostasis, the epicenter of the body's metabolic capability, a massive filter
detoxifying the portal blood releasing cleansed blood to the systemic circulation,
and a lymphoid organ protecting against infection. However high our regard for the
liver, it is dwarfed by the perspective of ancient civilizations that regarded the
liver as the seat of life and window to the future. In ancient Mesopotamia and Babylonia,
the liver was used to divine the future using a technique termed hepatoscopy. This
interest is captured in a Biblical quote from Ezekiel 21 : 21, “For the king of Babylon
stands at the parting of the way, at the head of the two ways, to use divination;
he shakes the arrows, he consults the household idols, he looks at the liver.” Hepatoscopy
was continued by the Greeks, Etruscans, and the Romans. The practice is continued
in a fashion today. Liver injury or neoplasia foretells a poor future for many pharmaceuticals
in development.
The liver plays a central role in processing dietary carbohydrates, lipids, amino
acids, and vitamins; in the synthesis and turnover of most plasma proteins; and in
the detoxification and biliary excretion of endogenous wastes and xenobiotic compounds.
The liver also functions as an important organ of the innate immune system, integrated
into the complex system of defense against foreign macromolecules. As such, hepatic
disorders have far-reaching consequences, given the dependence of other organs on
the metabolic function of the liver.
Origin, structure, and function
The embryonic origin of the liver is an out-pouching of the embryonic endoderm forming
the duodenum termed the hepatic diverticulum or the liver bud. Primitive epithelial
cells of the hepatic diverticulum extend into the adjacent mesenchymal stroma of the
septum transversum, a sheet of cells that incompletely separates the pericardial and
peritoneal cavity and that will develop into the connective tissue of the liver. The
primitive epithelial cells are arranged in close approximation with the vessels that
form the vitelline venous plexus, a complex of vessels that drain the yolk sac. Thus
the essential sinusoidal arrangement of the liver is established very early in development.
The gallbladder and the cystic duct arise from the caudal part of the hepatic diverticulum.
The hepatic diverticulum is also the origin of the biliary epithelium. Development
of the biliary tree begins at the hilus and spreads outward to reach the subcapsular
zone over time. Intrahepatic bile ducts develop from the ductal plate, a structure
that is composed initially of a single row of hepatoblasts that surround the portal
vein branches and ensheath the mesenchyme of the primitive portal tract. The development
of the hepatoblasts adjacent to the portal tract mesenchyme is altered by interactions
with the mesenchyme. Initially, these cells form the ductal plate, a single row of
cells surrounding the portal tract. The cells of the ductal plate can be identified
by expression of cytokeratin 7, differentiating them from the hepatoblasts (Fig. 2-1
). A second discontinuous outer layer of cells forms subsequently, and the 2-cell–thick
regions remodel into tubules. Most undergo apoptosis, but 1 or 2 ducts become incorporated
in the developing portal tract. At about the time the bile ducts are forming, the
hepatic artery branches appear in the developing portal tract. Nerves and lymphatics
eventually invest the portal tracts as well, completing the mature portal tract. The
wave of development from the hilus to the subcapsular region is imperfect, as the
veins, arteries, and bile ducts reach their terminal ends separately. Consequently,
in human liver, up to 30% of subcapsular portal tracts are “dyads” containing only
bile duct and artery profiles. Sinusoidal lining cells other than the endothelium
likely arise in the bone marrow and populate the liver via a hematogenous route.
Figure 2-1
The ductal plate of a fetal dog with 2-cell–thick rows of cytokeratin 7–positive cells
formed at the edge of the developing portal tract.
The liver is the largest internal organ in the body. In adult carnivores, the liver
constitutes ~3% of body weight. In adult omnivores, it is ~2% of body weight and ~1%
of body weight in adult herbivores. In neonates of all species, the liver is a larger
percentage of body weight than in the adult. The liver has a smooth capsular surface,
and the parenchyma consists of friable red-brown tissue that is divided into lobes.
The number and shape of the liver lobes of the major domestic mammals vary among species.
In monogastric animals, the liver abuts the diaphragm and occupies the central area
of the cranial abdomen. In ruminants, and to a lesser extent in horses, the liver
is displaced to the right side of the cranial abdominal cavity. A series of ligaments
maintains the liver in its position. The coronary ligament attaches the liver to the
diaphragm near the esophagus. The falciform ligament attaches the midline of the liver
to the ventral midline of the abdomen. The round ligament, a remnant of the umbilical
vein, is embedded within the falciform ligament.
The liver receives ~25% of the cardiac output, but is also supplied by the portal
vein. The valveless portal vein drains the digestive tract, forestomachs, glandular
stomach, and intestines, as well as the spleen and pancreas. Portal vein flow provides
70-80% of the total afferent hepatic blood flow and ~50% of the oxygen supply. The
hepatic artery provides the remainder of hepatic blood flow. Portal blood flow is
not regulated, but hepatic arterial flow is regulated, primarily by adenosine, and
responds to changes in portal flow. When portal blood flow is reduced, less adenosine
is washed out of the hepatic circulation, and this increased concentration drives
hepatic arterial dilation, creating a hepatic arterial buffer effect whereby a consistent
hepatic blood flow is maintained. Portal blood flow is important for the rapid clearance
of nutrients, xenobiotics, microorganisms, and potentially immunogenic materials that
enter the circulation from the gastrointestinal tract. Hepatic arterioles disperse
into a peribiliary capillary plexus, a perivenous plexus surrounding the portal vein,
or join terminal hepatic arterioles before entering the sinusoids, lowering pressure
and preventing reversal of portal venous inflow. Portal and arterial blood eventually
mix in the low-pressure hepatic sinusoids. Both terminal portal venules and hepatic
arterioles flow into the sinusoids, but flow is closely regulated by a series of inlet
sphincters formed by endothelial cells for the venules and smooth muscle for the arterioles.
Thus, at any particular time, sinusoidal blood could be entirely venous, mixed arterial
and venous, or arterial. This blood flow pattern may account for the interlobular
and intralobular heterogeneity of lesions following various toxicities. Blood leaves
the liver via the hepatic vein, which is very short, and enters the caudal vena cava.
Hepatic sinusoids have an average diameter of 10 µm, but can expand up to 30 µm. The
periportal sinusoids are more tortuous than those in the centrilobular region. Hepatic
sinusoids are lined by specialized endothelial cells. Hepatic sinusoids differ from
vascular structures elsewhere in that they lack a typical basement membrane, and are
supported by a specialized, discontinuous or loose extracellular matrix (ECM). Hepatic
sinusoidal endothelial cells are fenestrated, and these 100-nm diameter sieve-like
pores control fluid, solute, and particulate interchange between blood and the perisinusoidal
space, regulated by the action of the cellular cytoskeleton. Sinusoidal endothelial
cells are actively pinocytotic and internalize and degrade various endogenous glycoproteins,
glycosaminoglycans, and immune complexes. Kupffer cells are fixed macrophages attached
to the inner sinusoidal wall in direct contact with blood moving at a relatively low
velocity. This arrangement facilitates phagocytic removal of particulates, especially
bacteria that enter the portal blood via the lower alimentary tract. Kupffer cells
also participate in the regulation of inflammatory and repair responses by secretion
of various cytokines into the circulation and perisinusoidal space. Natural killer
cells (formerly referred to as pit cells) are large granular lymphocytes with natural
killer activity that adhere to the sinusoidal endothelium, where they are also well
situated to participate in various innate immune defenses, for example, targeting
infected cells that enter the liver via the blood.
The sinusoids are separated from the adjacent hepatocellular plates by an extracellular
space, known as the space of Disse, that contains hepatic stellate cells (also termed
lipocytes or Ito cells), reticulin fibers, and nerves. The space of Disse is not readily
visible by light microscopy unless there is fluid retention, such as can occur with
impediment to venous outflow. Although hepatic sinusoids in the normal liver lack
a conventional basement membrane, the perisinusoidal space contains a low-density
ECM consisting of collagen type IV; laminin; fibronectin; minor amounts of collagen
types I, III, V, and VI; nonfibrillar collagen XVIII; tenascin; and various proteoglycans.
A conventional ECM composed of fibrillar collagen types I, III, V, and fibronectin
is found in the external capsule (Glisson's capsule), septa, and around portal triads
and central veins. “Reticulin fibers” are the components of the ECM that are stainable
by silver impregnation techniques, consisting mainly of collagen type III with attached
fibronectin and other glycoproteins. The fenestrated sinusoidal endothelium, coupled
with the loose subendothelial matrix, allows for exchange of various macromolecules
between hepatocytes and the sinusoidal blood. After hepatic injury, a denser, less
permeable matrix resembling a true basement membrane may form, and sinusoidal endothelial
cells may lose their fenestrae (so-called “capillarization” of sinusoids), reducing
uptake and secretion of plasma proteins and other metabolically important substances.
The terminal hepatic venules
(“central veins”) collect the outflow blood from the sinusoids. These venules converge
into the larger hepatic veins that empty into the caudal vena cava. In most species,
increased pressure in the vena cava during right-sided heart failure or hepatic vein
thrombosis causes passive congestion and distension of the hepatic veins and sinusoids.
However, the large and small hepatic veins in dogs have a prominent spiral circumferential
smooth muscle that can also affect the central venous pressure on the sinusoids. Fluids
from the perisinusoidal space drain into lymphatics in the extracellular connective-tissue
spaces of the liver capsule, the portal tracts, and the connective tissue of the terminal
veins. These flow out the portal hilus to the hepatic lymph nodes and eventually enter
the thoracic duct. In some species, such as the dog, there are also lymphatics around
the larger hepatic veins; these cross the diaphragm into the mediastinum. The liver
is the largest lymph producer in the body, contributing 20-50% of the thoracic duct
flow. Hepatic lymph is high in protein, containing 85-95% of the protein of plasma
and a high cell count composed of lymphocytes and macrophages. In sheep, more lymphocytes
pass through the liver than any typical lymphoid organ, and ~2 × 108 macrophages leave
the liver in lymph daily. Hepatic nerves contain both sympathetic and parasympathetic
fibers. The fibers invest major blood vessels and also extend along the sinusoids.
These may modulate function of hepatocytes, endothelial cells, and hepatic stellate
cells.
The vasculature of the liver parenchyma defines its functional microanatomy, but debate
continues as to what best represents the hepatic structural-functional unit. Mammalian
hepatocytes are organized in plate-like monolayer arrays among the sinusoids and in
3 dimensions; plates, sinusoids, and tracts anastomose in a complex pattern. Currently,
a somewhat baffling array of models exists, each with their own adherents. These include
the well-known lobular and acinar patterns (discussed later) and several others. Matsumoto's
primary lobule is based on detailed reconstructions of human liver sections, and considers
the penetrating venule extending from the portal tract as a “vascular septum” and
the origin of the primary lobule's blood flow as it is a starting place for the radially
arranged sinusoids flowing to the terminal hepatic vein. In this model, there is a
series of branches formed by the portal vein. The first branches provide a conducting
portal flow, and the next level of branches drain directly into the sinusoids forming
the distributing portal flow. The choleohepaton, related to the concept of the nephron,
is composed of an isosceles triangle of hepatocytes with its apex in contact with
the terminal hepatic venule and drained by a single bile ductule/canal of Hering at
the base of the triangle. More detail is available in the cited references.
Arrangements of hepatocytes in the most commonly used nomenclature systems are referred
to as either acini or lobules.
•
The classic hepatic lobule is a 6-sided anatomic arrangement of hepatocytes centered
on the terminal hepatic venule, also termed the “central vein” in this context. Peripherally,
lobules are outlined by fibrovascular septa extending from the portal tracts. In the
pig liver, septa form obvious lobular perimeters, but in most mammalian species, the
lobules are less pronounced because connective tissue is more restricted to portal
tracts. The terms periportal and centrilobular are mainly used for pathologic conditions
that are centered on the hepatocytes surrounding the portal tracts or the central
veins of the classic lobule.
•
The hepatic acinus of Rappaport is a functional diamond-shaped subunit divided into
zones in relation to blood supply:
•
Zone 1 hepatocytes are arranged around an axis formed by the portal tract and the
distributing vascular branches that leave the portal tract and are closest to the
oxygen- and nutrient-rich arterial and portal inflow.
•
Zone 2 is the transitional midzone.
•
Zone 3 (periacinar) hepatocytes form the apex of the diamond-shaped acinus, are nearest
the outflow (terminal hepatic venule), and are exposed to reduced oxygen and nutrients.
The functional activity of hepatocytes is heterogeneous, and virtually all liver functions
have a zonal gradient. Periportal hepatocytes, exposed to the blood with the highest
concentration of oxygen, insulin, glucagon, and amino acids, are the principal site
of gluconeogenesis, protein synthesis, aerobic metabolism, urea cycle, and lipid and
cholesterol metabolism. In the centrilobular region, glycolysis, lipogenesis, and
the major biotransformation functions are more active, including the expression of
most cytochromes P450, glucuronyl transferases, glutathione S–transferases, and other
biotransformation/detoxification enzymes. Centrilobular hepatocytes are therefore
more susceptible to hypoxic injury as well as injury by toxic substances that are
metabolically activated by cytochromes P450. By comparison, hepatocytes in periportal
hepatocytes are more susceptible to direct-acting toxicants, such as ingested metal
salts, given their proximity to the vascular inflow. Under the influences of various
inducers, the patterns of enzyme expression can extend beyond the resting limits.
Lobular variation is not restricted to parenchymal cells, but is also apparent in
the structure and function of sinusoidal endothelial cells, Kupffer cells, perisinusoidal
stellate cells, and the composition of the matrix in the space of Disse.
The portal tract, or portal triad, is a well-defined structure containing at least
one small arterial branch, a portal vein branch, and a bile duct, surrounded by connective
tissue composed primarily of type I collagen (Fig. 2-2
). Because of the pattern of progressive branching of the portal tract system, individual
tracts exhibit a range of sizes and shapes, from round to triangular or branching.
In larger portal tracts, lymphatic channels and autonomic nerve fibers may be seen.
The bile duct system is a branching outflow that ultimately enters the proximal duodenum.
Most species, with the exception of the horse and rat, have a bile storage diverticulum
(gallbladder). Cats occasionally have divided or bipartite gallbladders. The bile
duct joins the pancreatic duct before entry into the duodenum in some species and
has a separate entry in others. Intrahepatic bile ducts range in size from the larger
septal or trabecular ducts (internal diameter of >100 µm in humans) to the smaller
interlobular ducts, and tend to be adjacent to a hepatic artery branch of approximately
the same size. Bile ducts are lined by cuboidal to low columnar bile duct epithelial
cells, subtended by a periodic acid–Schiff (PAS)-positive basement membrane. Bile
ductules are smaller yet (lumen size of <20 µm) and are located at the periphery of
portal tracts.
Figure 2-2
The normal portal tract contains a branch of the portal vein and hepatic artery, as
well as a bile duct. The first row of hepatocytes adjacent to the portal tract connective
tissue is termed the limiting plate.
Cells of the liver
Hepatocytes (referred to as parenchymal cells) constitute ~70-80% of the liver mass.
However, >50% of liver DNA is found in smaller nonparenchymal cells (bile duct epithelium,
hepatic stellate cells, sinusoidal endothelium, Kupffer cells) and itinerant cells
(such as leukocytes). The hepatocyte is a polygonal epithelial cell, ~30-40 µm in
diameter, arranged in single-cell–thick anastomosing plates, separated by hepatic
sinusoids. Each hepatocyte is therefore exposed to sinusoidal blood on 2 sides. A
discontinuous line of hepatocytes, termed the limiting plate, is found at the interface
with the collagenous ECM of the portal tract. Normal hepatocytes have abundant eosinophilic
cytoplasm, and most have a single, round, centrally placed nucleus with finely dispersed
chromatin and at least one nucleolus. Some binucleate hepatocytes are present normally
in mammals and can become more numerous in response to various stimuli and injuries
that induce or affect regeneration.
Hepatocytes are metabolically highly active cells, containing an array of organelles,
including smooth and rough endoplasmic reticulum, mitochondria, lysosomes, peroxisomes,
Golgi complexes, and transport vesicles. These organelles support a variety of hepatocellular
functions, including the synthesis and secretion of plasma proteins, coagulation factors,
and acute-phase proteins. Hepatocytes store nutrients in times of adequate energy
and release glucose when needed. They are key modulators of lipid metabolism, and
they synthesize and secrete lipoproteins. In addition, they are the only cells capable
of bile acid synthesis, and they can absorb and secrete them into bile. Finally, hepatocytes
detoxify the large majority of xenobiotics and secrete them into the bile. Because
of this central role in metabolism, the liver is subjected to a variety of nutritionally
based insults as well as toxin-related damage. A greater proportion of the genome
is expressed in the normal liver than has been observed in any other tissue, an indication
that brief surveys of liver functions are necessarily oversimplified. However, those
constituents that are most abundant have most influence on the microscopic appearance
of the liver. The normal hepatocyte contains abundant glycogen, which varies depending
on food intake, and which can be demonstrated by PAS staining, as well as variable
amounts of stored triglycerides and various proteins, such as ferritin, an iron-binding
protein. The cytoplasm of the centrilobular hepatocytes may also contain uniform,
golden-brown granules of lipofuscin, particularly in older cats. This so-called “wear-and-tear”
pigment becomes more prominent with age, and progressively accumulates in midzonal
and periportal hepatocytes. Hepatocytes have a cytoskeleton composed of microtubules,
microfilaments, and intermediate filaments. Microtubules are found throughout the
cytoplasm, and are involved in the movement of secreted proteins into the extracellular
perisinusoidal space; accordingly, microtubule inhibitors such as colchicine and Vinca
alkaloids may reduce hepatic protein secretion. Microfilaments, composed of actin
and myosin, are concentrated around the bile canaliculus, where they are involved
in canalicular peristalsis and bile secretion; microfilament inhibitors result in
cholestasis. Intermediate filaments (predominantly cytokeratins 8 and 18) form an
irregular meshwork extending from the plasma membrane to the perinuclear zone, and
are responsible for spatial organization of the hepatocyte.
There are 3 morphologically and functionally distinct surfaces of the hepatocyte plasma
membrane.
1.
The sinusoidal domain faces the space of Disse and has numerous irregular microvilli,
increasing hepatocyte surface area by ~6-fold (considerably less that that seen in
enterocytes). This specialized membrane is modified to facilitate an exchange of substances
with the blood. There are ultrastructurally evident pits between the villi, some of
which represent secretory vacuoles in the process of exocytosis, sending various products
into the plasma, and others are clatharin-coated pits involved in selective receptor-mediated
endocytosis. Numerous membrane receptors for glycoproteins, asialoglycoproteins, peptides,
hormones, growth factors, immunoglobulin A, and other endocytotic or signaling ligands
are found at the sinusoidal pole. In addition, transmembrane proteins involved in
plasma exchange of small ionic substances with the sinusoidal plasma, and transmembrane
proteins responsible for matrix recognition, are concentrated on the sinusoidal surface.
2.
The lateral domain extends from the sinusoidal surface to the edge of the canaliculus.
This portion of the cell membrane is specialized for adhesion via junctional complexes,
including desmosomes, tight junctions, and intermediate junctions, as well as for
intercellular communication via gap junctions.
3.
The canalicular domain is the beginning of the bile drainage system of the liver.
The canaliculus is an intercellular space between 2 adjacent hepatocytes, isolated
by junctional complexes. The canalicular surface is covered with an irregular array
of microvilli. Canalicular diameter increases as it approaches the periportal region,
enlarging from ~0.5-2.5 µm. Bile is propelled along the canaliculi by a web of contractile
microfilaments. This specialized membrane contains various adenosine triphosphate
(ATP)-dependent carriers that export many products, including leukotrienes, bile salts,
xenobiotics, and their metabolites into the bile.
The canal of Hering is partly lined by biliary epithelium and partly by hepatocytes
and connects the bile canaliculus to the cholangioles and eventually the interlobular
bile ducts.
Cholangiocytes (biliary epithelium) account for ~3-5% of the liver cell population.
Although derived from common embryologic progenitor cells, cholangiocytes differ from
hepatocytes in both phenotype and function. They contain a strongly developed network
of intermediate filaments, including cytokeratins 7 and 19. They also express marked
heterogeneity along the anatomic course of the biliary system. Functionally, bile
duct epithelial cells actively modify the composition of bile. Secretion is primarily
under the control of secretin and somatostatin. Secretin released from the duodenum
triggers secretion of bicarbonate-rich fluids that buffer acids released from the
stomach. Cholangiocytes secrete immunoglobulin A (IgA) and IgM, but not IgG. Absorption
involves the sodium-dependent glucose transporter and aquaporins responsible for glucose
and water uptake, as in the renal proximal tubule. They express γ-glutamyltranspeptidase,
which removes glutamic acid from glutathione conjugates.
Hepatic progenitor cells (HPCs), formerly known as oval cells in rodents, reside in
the region of the canal of Hering. These cells are bipotential and can mature into
either biliary epithelium or hepatocytes. They can proliferate and form a type of
ductular reaction. In cases of massive hepatic necrosis in which the animal survives
for a few days after the initial injury, HPCs can proliferate dramatically, forming
cords or small caliber ducts lined by cuboidal basophilic cells with abundant mitochondria.
These cells can mature and replace lost hepatocytes and bile ducts. HPCs in humans
and rats contain both markers of hepatocyte phenotype (i.e., albumin) and biliary
phenotype (i.e., cytokeratin 7). Similar markers have been described in dogs and cats
as well. Bone marrow–derived pluripotential stem cells also appear to have the ability
to differentiate into hepatocytes. Other types of ductular reaction are discussed
in the section on Responses of the liver to injury.
Hepatic endothelial cells are specialized, perforated by numerous fenestrations, ~175 nm
in diameter, and often clustered together forming sieve plates. Larger, but less frequent,
fenestrations are more common at the periportal end of the sinusoid, but opening size
is dynamic, responding to endogenous mediators and toxins. The endothelial cells rest
on a very thin and discontinuous ECM. The fenestrations allow direct contact between
the sinusoidal lumen and the space of Disse. Only larger particles, such as chylomicrons,
and cells are excluded. Sinusoidal endothelial cells differ from normal vascular endothelium
in several additional ways, including the absence of factor VIII–related antigen (except
in inflammatory conditions) and high endocytic activity. Endocytosis of immune complexes
and some proteoglycans are major functions. They also synthesize molecules that affect
vascular tone such as nitric oxide, endothelins, and prostaglandins.
Kupffer cells
are specialized macrophages located in sinusoidal lumens, mainly at branch points.
Once thought to be “fixed,” it is now known that they can migrate along the sinusoid
and into areas of tissue injury (Fig. 2-3
). Kupffer cells may have a dual origin as they are derived, at least in part, from
blood-borne monocytes, but they are also capable of local proliferation, particularly
in inflammation. They are not efficient antigen presenters, but they are proficient
phagocytes of apoptotic and necrotic cells, particulates, and microorganisms; consequently,
the liver is a major “filtering organ” for the body. There are species differences
in the efficiency of this process. Clearance of particulates, and endotoxin in particular,
is accomplished more effectively by Kupffer cells in dogs, humans, and laboratory
rodents than in ruminants, horse, pig, cats, and whales, species that have a significant
population of intravascular macrophages in the pulmonary vasculature. Kupffer cells
can phagocytose a variety of gut-derived materials; bacteria, various biologically
active bacterial components, including lipopolysaccharides, lipoteichoic acids, and
peptidoglycans, without stimulating inflammation. Activated Kupffer cells can secrete
tumor necrosis factor-α and other cytokines, and nitric oxide; these contribute to
peripheral vasodilation and hypotension in systemic inflammatory response syndromes
initiated by bacterial components. Other secreted cytokines, such as interleukin-1
(IL-1) and IL-6, mediate the acute-phase response and some aspects of the immune and
liver regenerative responses. However, there can be a balance in proinflammatory and
anti-inflammatory signaling as there are distinct differences in the signaling repertoire
of different Kupffer cells. Some Kupffer cells are more likely to secrete IL-10, which
can suppress macrophage activation and cytokine secretion. Cytokine responses of Kupffer
cells are believed to be important in regulating the extent of adaptive immune response
or tolerance to potentially antigenic macromolecules that can reach the liver through
the portal blood.
Figure 2-3
Kupffer cells, stained with antibodies against myeloperoxidase, line the sinusoids
at regular intervals.
The liver contains large numbers of lymphocytes, comprising ~5% of the entire cell
population of the liver, with an organ-specific lymphocyte distribution characterized
by the enrichment of elements of the innate immune system, including natural killer
T lymphocytes (NKT cells), natural killer (NK cells), and innate lymphocytes, in addition
to the Kupffer cells previously mentioned. Elements of the acquired immune system,
CD8+ T cells, are also increased when compared with peripheral blood. The majority
of intrahepatic lymphocytes are involved in innate immune responses rather than acquired
immunity. Hepatic NK cells constitute ~40% of hepatic lymphocytes and are distinct
phenotypically and functionally from blood NK cells. Hepatic NKT cells reside in the
space of Disse, are considered large granular lymphocytes, and were previously referred
to as pit cells. Intrahepatic NK cells have important functions in defense against
foreign antigens released from the gut, viral infections, metastatic tumors, hepatocellular
carcinoma, and modulation of hepatic fibrosis. The liver also contains the largest
population of γδ T cells in the body. Although the precise function of these diverse
lymphocyte types is not currently understood, their large number alone suggests that
they must be involved in immunologic homeostasis and respond to immunologic challenges,
indicating that the liver can be considered a lymphoid organ.
Hepatic dendritic cells play an important role in the induction and regulation of
immune responses. These antigen-presenting cells have only recently been studied in
the liver. There are several other antigen-presenting cells in the liver, including
the sinusoidal endothelial cells and Kupffer cells. Unlike cells that reside within
the sinusoid, hepatic dendritic cells are found within the portal tract. Hepatic dendritic
cells are considered to be functionally immature compared to the dendritic cells of
the spleen and bone marrow, and they may be involved in immune tolerance in the liver.
Hepatic stellate cells (HSC), originally described by Boll and von Kupffer in the
1870s, were neglected until the 1950s, when they were described in detail by Ito.
They have also been known as lipocytes, Ito cells, or fat-storing cells. HSC reside
in the space of Disse, but there are other populations of similar cells with the ability
to produce ECM and to transform into a myofibroblast phenotype within the connective
tissue of the portal tract and centrilobular veins. There are 4 key functions of HSC:
(1) storage and homeostasis of retinoids, including vitamin A; (2) maintenance and
remodeling of the sinusoidal ECM in health and disease; (3) production of growth factors,
such as hepatocyte growth factor and various cytokines; and (4) regulation of sinusoidal
diameter by contraction of cellular processes. This may be in response to adrenergic
stimulation, as all HSC are in contact with autonomic nerve fibers. HSC can become
greatly distended with lipid in carnivores on some diets.
Activation of HSC has been extensively studied because of their importance in hepatic
fibrosis. In the transition from quiescence to activation, HSC lose their characteristic
lipid droplets, possibly catabolizing the lipid to support their activation. They
then develop a myofibroblast phenotype characterized by the expression α–smooth muscle
actin. Proinflammatory cytokines released primarily by Kupffer cells, such as transforming
growth factor-β released in response to tissue injury, stimulate HSC to increase the
deposition of ECM, including collagen type I, III, and IV, and laminin. This new ECM
transforms the sinusoid to a less permeable capillary, lined by a basement membrane–like
layer and without fenestrations in the sinusoidal endothelium, reducing transfer of
macromolecules between hepatocytes and the blood. The acquired contractility of the
HSC during fibrogenesis is increased because of an increase in the contractile stimulus
of endothelin-1 and a reduction in vasodilation driven by diminished nitric oxide
generation. In severe chronic injury leading to cirrhosis, the increased expression
of contractile proteins within activated stellate cells can further restrict sinusoidal
blood flow as a primary effect, rather than a consequence of nodule formation and
fibrosis. Peribiliary fibrosis arises from activation of the circumferential fibroblasts
of the bile ducts that undergo a phenotypic transformation similar to that of the
HSC along the sinusoids. Fibrosis of the portal tracts and the central vein connective
tissue develops from activation of myofibroblasts resident in these areas as well.
It is also possible that epithelial-mesenchymal transition of hepatocytes, biliary
epithelial cells, or HSC can contribute to hepatic fibrosis during chronic injury.
Mast cells are abundant in the liver, particularly in dogs. They typically occupy
a perivenous location, where they may influence vascular tone and respond to various
potentially injurious substances or organisms. Degranulation of mast cells in the
liver leads to contraction of the spiral smooth muscle, restricting blood outflow
from the canine liver. This is a feature of shock in dogs.
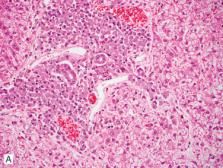
Hematopoiesis in the fetal life of mammals occurs mainly in the perisinusoidal compartment
of the liver sinusoids. Postnatally, hepatic hematopoiesis declines but can return
as extramedullary hematopoiesis in conditions of increased demand (Fig. 2-4
). Because the liver is an early site of hematopoiesis, the environmental conditions
and cells, including resident populations of appropriate stromal cells and, possibly
hematopoietic stem cells, remain supportive for the initiation or reactivation of
a stem cell niche. The degree of hepatic extramedullary hematopoiesis in larger species
can have diagnostic significance, but in laboratory rodents and other small animals,
it can be an incidental observation.
Figure 2-4
Hepatic extramedullary hematopoiesis in a dog.
Further reading
Crawford JM, Burt AD. Anatomy, pathophysiology and basic mechanisms of disease. In:
Burt AD, et al., editors. Macsween's Pathology of the Liver. 6th ed. New York: Churchill
Livingstone; 2012. p. 2-77.
Desmet VJ. Ductal plates in hepatic ductular reactions. Hypothesis and implications.
I. Types of ductular reaction reconsidered. Virchows Arch 2011;458:251-259.
Desmet VJ. Ductal plates in hepatic ductular reactions. Hypothesis and implications.
III. Implications for liver pathology. Virchows Arch 2011;458:271-279.
Dixon LJ, et al. Kupffer cells in the liver. Compr Physiol 2013;3:785-797.
Ekataksin W, Wake K. The anatomy and physiology of the liver. In: Boyer JL, Ockner
RK, editors. Progress in Liver Disease. Philadelphia: WB Saunders; 1997. p. 1-30.
Johns JL, Christopher MM. Extramedullary hematopoiesis: a new look at the underlying
stem cell niche, theories of development, and occurrence in animals. Vet Pathol 2012;49:508-523.
Malarkey DE, et al. New insights into functional aspects of liver morphology. Toxicol
Pathol 2005;33:27-34.
Matsumoto T, Kawakami M. The unit-concept of hepatic parenchyma—a re-examination based
on angioarchitectural studies. Acta Pathol Jpn 1982;32(Suppl. 2):285-314.
Oda M, et al. Regulatory mechanisms of hepatic microcirculatory hemodynamics: hepatic
arterial system. Clin Hemorheol Microcirc 2006;34:11-26.
Winkler GC. Pulmonary intravascular macrophages in domestic animal species: review
of structural and functional properties. Am J Anat 1988;181:217-234.
Yamamoto K. Electron microscopy of mast cells in the venous wall of canine liver.
J Vet Med Sci 2000;62:1183-1188.
Developmental Disorders
Hepatic cysts
Serous cysts are occasionally found attached to the capsule on the diaphragmatic surface
in calves, lambs, and foals (Fig. 2-5
). These cysts are usually small and multiple, but some are isolated and very large.
Cyst walls are composed of connective tissue lined by flattened or cuboidal epithelium.
The content is clear and serous. Their origin is not known, but it is variously postulated
that they are serosal inclusion cysts, part of congenital polycystic biliary anomalies,
or of endodermal origin. They do not contain bile. The declining incidence of these
anomalies with age suggests that a large proportion of them involute in the early
postnatal period.
Figure 2-5
Serous cyst attached by a stalk to the hepatic capsule in a 3-day-old Holstein calf.
(Courtesy J.L. Caswell.)
Solitary biliary cysts—single round cysts lined by a flattened single layer of biliary
epithelium—are uncommon and may be congenital or acquired. Multiple hepatic peribiliary
cysts putatively arising from peribiliary glands have been reported in a 6-month-old
pig.
Hamartomas
Von Meyenburg complexes (biliary microhamartomas) are developmental malformations
arising from persistent embryonic ductal plate remnants. These are discrete, usually
subcapsular, fibrotic areas containing small, irregularly shaped, often dilated, U-shaped
or branching, bile duct–like structures lined by low cuboidal epithelium (Fig. 2-6
).
Figure 2-6
Von Meyenburg complexes in a dog liver.
Mesenchymal or mixed liver hamartomas, rare benign tumor-like lesions characterized
by disorganized hepatocellular and/or biliary structures embedded in a mucinous primitive
mesenchyme have been reported in 2 equine fetuses.
Ductal plate malformations
Persistence and/or aberrant remodeling of the embryonic ductal plate can give rise
to a spectrum of cystic biliary diseases. Congenital hepatic fibrocystic diseases,
part of the group of hepatorenal fibrocystic disease that includes the polycystic
kidney diseases, are a product of ductal plate malformations occurring at different
levels of the biliary tree. Analysis of the underlying genetic basis of the human
hepatorenal fibrocystic diseases has identified defective protein components in primary
cilia and associated basal bodies. These mechanotransducer organelles are involved
in environmental monitoring, signal transduction, and cell proliferation, and are
important in the normal development of the biliary system in the liver, as well as
renal tubules. As such, many of these diseases are now considered “ciliopathies.”
In human hepatopathology, the hepatorenal fibrocystic diseases can be grouped into
3 descriptive categories: (1) polycystic liver disease (often seen in association
with autosomal dominant polycystic kidney disease of adults), characterized by isolated
microscopic to macroscopic unilocular or multilocular cysts in a fibrous stroma, with
no continuity with the intrahepatic biliary tree, thought to originate from von Meyenburg
complexes in the most peripheral branches of the biliary tree; (2) congenital hepatic
fibrosis (often seen in association with autosomal recessive polycystic kidney disease
of childhood), characterized by defective remodeling of the ductal plate at the level
of interlobular ducts, with excess abnormally shaped embryonic bile ducts retained
in the primitive ductal plate configuration, abnormal portal veins, and progressive
fibrosis of the portal tracts; and (3) Caroli disease, characterized by non-obstructive
saccular or fusiform dilation of medium- and large-sized intrahepatic bile ducts,
with maintenance of continuity with the biliary system. Caroli syndrome refers to
Caroli disease co-occurring with congenital hepatic fibrosis. In veterinary medicine,
a similar classification of the liver lesions has been proposed: adult polycystic
disease (including von Meyenburg complexes), juvenile polycystic disease/congenital
hepatic fibrosis, and congenital dilation of the large and segmental bile ducts (resembling
Caroli disease).
Congenital cystic lesions involving the hepatic biliary system and kidneys have been
reported in juvenile dogs, cats, pigs, goats, and foals, and have been compared to
congenital hepatorenal fibrocystic disorders of humans. Cysts may also be found in
the pancreas or other organs. Animals may die from progressive renal insufficiency,
and/or from hepatic dysfunction and portal hypertension associated with hepatic fibrosis.
Hepatic fibrosis and cysts are present in a significant proportion of cats with polycystic
kidney disease, inherited in Persian cats, exotic shorthaired, and other related breeds
as an autosomal dominant C→A transversion mutation in exon 29 of the feline PKD1 gene,
resembling the adult form of polycystic kidney disease in humans. The liver lesions
have been more difficult to classify, and may appear as multiple large cysts resembling
adult-type polycystic disease, as congenital hepatic fibrosis characterized by portoportal
bridging fibrosis with excess abnormally formed bile ductules (Fig. 2-7
), or as combinations of both lesions. Polycystic kidney and liver disease reported
in West Highland White and Cairn Terrier litters resembles the autosomal recessive
polycystic kidney disease of children. Congenital hepatic fibrosis has been described
in dogs. Affected animals are presented at or before a year of age with clinical signs
of liver disease, including ascites, microhepatica, and extrahepatic portosystemic
shunts. Histologically, these dogs had livers with extensive bands of portal bridging
fibrosis containing numerous small irregular, tortuous bile ducts, often accompanied
by absent or hypoplastic portal veins and compensatory arteriolar proliferation, and
with no evidence of nodular regeneration and minimal inflammation, allowing differentiation
of this congenital condition from acquired chronic liver disease. Congenital hepatic
fibrosis has also been reported in aborted and neonatal calves, in the latter case
accompanied by cyst formation in the kidney and lung. Congenital hepatic fibrosis
with cystic bile ducts has been described in Swiss Freiberger foals (Fig. 2-8
) and is seen occasionally in other breeds, with generalized portal bridging fibrosis
containing many small, irregularly formed and occasionally cystic bile ducts. Macroscopic
congenital dilation of the large and segmental bile ducts and diffuse cystic kidney
disease, resembling Caroli disease, has been reported in dogs.
Figure 2-7
Congenital hepatic fibrosis in a Himalayan cat with polycystic kidney disease.
Figure 2-8
Congenital hepatic fibrosis in a Swiss Freiberger foal.
Extrahepatic biliary anomalies
Cats occasionally have divided or bipartite gallbladders. Reduplication of the gallbladder
has also been reported in swine. Other anomalies of the extrahepatic biliary system
include agenesis of the gallbladder reported in dogs, and absence or atresia of one
or more ducts, reported in lambs, calves (eFig. 2-1), foals, a cat, a dog, and a pig.
In carnivores, bile duct atresia may lead not only to jaundice, but also to vitamin
D–deficiency rickets, because of their inability to absorb fat-soluble vitamins. Congenital
atresia may be associated with defects in the developmental morphogenesis of bile
ducts, or in utero vascular, inflammatory, or toxic insults to the biliary tree that
culminate in the obliteration of the lumen.
eFigure 2-1
Congenital biliary atresia and gallbladder agenesis in a calf.
(Courtesy B. Njaa.)
Choledochal cysts arising from the cystic or common bile duct have been described
in cats.
Congenital vascular anomalies
These include congenital portal vein aneurysms, hepatic arteriovenous malformations,
congenital portosystemic shunts between the portal vein and other systemic veins,
and primary hypoplasia of the portal vein. Extrahepatic
congenital
portosystemic shunts are readily distinguished from shunts that are
acquired
during portal hypertension, as acquired shunts are typically multiple, thin-walled,
tortuous collateral venous connections between the portal vein or its tributaries
and caudal vena cava, renal vein, or azygos vein (Fig. 2-9
). Although multiple acquired shunts do not develop in the presence of congenital
PSS, they can arise with other congenital abnormalities, such as arteriovenous malformations
or hypoplasia or dysplasia of portal veins, as a consequence of portal hypertension.
Acquired shunts resulting from portal hypertension secondary to liver injury and repair
are discussed later in the section Vascular factors in hepatic injury and circulatory
disorders.
Figure 2-9
Multiple acquired portosystemic vascular shunts in a dog with chronic liver disease.
Portal vein aneurysms, both congenital and acquired as a consequence of concurrent
liver disease, have been described in dogs. Extrahepatic aneurysms were always located
at the level of the gastroduodenal vein insertion. All were asymptomatic, although
predisposed to portal vein thrombosis.
Hepatic arteriovenous malformations have been reported in dogs and cats. These are
congenital or, in some instances, acquired communications between branches of the
hepatic artery, and more rarely, the gastroduodenal artery and left gastric artery
and portal vein. Mixing of higher-pressure arterial blood with venous blood results
in retrograde flow into the portal vein, arterialization of the portal circulation,
and development of portal hypertension, with the opening of vestigial, low-resistance,
collateral, extrahepatic portosystemic communications (acquired extrahepatic shunts).
The fistulae may be macroscopic or microscopic, are typically multiple, and may involve
one or more lobes of the liver. The hepatic parenchyma of affected lobes may be atrophied,
with dilated, tortuous, pulsatile vessels visible on the capsular surface. Histopathologic
findings include hyperplasia and anastomoses of arterioles and venules (Fig. 2-10
). Affected vessels have irregularly thickened walls with intimal hyperplasia consisting
of smooth muscle proliferation and deposition of elastin fibers, focal subintimal
fibromuscular proliferation, and smooth muscle hyperplasia of the tunica media. Degenerative
changes characterized by deposition of mucinous material and mineral in the intima
and media of arterioles, as well as thrombosis and recanalization of portal veins,
are also observed. Adjacent hepatic parenchyma may be atrophic, with periportal fibrosis,
and bile duct hyperplasia, arteriolar proliferation, and relative collapse of portal
vein branches within portal tracts. Arteriovenous fistulae may also be acquired, developing
subsequent to abdominal trauma, rupture of hepatic artery aneurysms, and secondary
to hepatic vein obstruction or cirrhosis with extreme portal hypertension.
Figure 2-10
Congenital intrahepatic arterioportal fistulae with thick-walled anastomosing vessels
and atrophy of adjacent parenchyma in a dog.
Congenital portosystemic vascular anomalies
are typically single anomalous vessels that directly connect the portal venous system
with the systemic venous circulation, bypassing the hepatic sinusoids and hepatic
parenchyma. They occur in dogs and cats, and, rarely, in pigs, foals, goats and calves.
These portosystemic shunts (PSS) may be either intrahepatic or extrahepatic in location.
The most common intrahepatic shunt, located in the left hepatic division, is a persistent
patent ductus venosus (Fig. 2-11
). Central and right divisional intrahepatic shunts have also been described in dogs
and cats. The major types of extrahepatic shunts include direct shunting from the
portal vein or major tributary (typically left gastric or splenic veins, less commonly
the gastroduodenal or mesenteric veins) to the caudal vena cava (portocaval shunt)
(Fig. 2-12
, eFig. 2-2) or to the azygos vein (portoazygos shunt), or connection of the portal
vein to the caudal vena cava, which itself shunts to the azygos vein. Extrahepatic
portosystemic shunts may also have hypoplasia of the portal vein distal to the origin
of the shunt. Large-breed dogs typically have intrahepatic shunts, usually a patent
ductus venosus, but sometimes other large intrahepatic communications. Small-breed
dogs and cats usually have single large extrahepatic shunts between the portal vein
and vena cava or azygos vein. An inherited basis is suspected for several breeds,
including Irish Wolfhounds, Maltese, Yorkshire Terriers, and Australian cattle dogs.
Figure 2-11
Congenital intrahepatic shunt, persistent patent ductus venosus in a dog.
(Courtesy J.L. Caswell.)
Figure 2-12
Congenital extrahepatic portocaval shunt in a cat.
eFigure 2-2
Congenital extrahepatic portocaval shunt in a dog.
(Courtesy University of Guelph.)
Affected dogs are usually presented in adolescence with failure to thrive or with
the neurobehavioral manifestations of hepatic encephalopathy. Often, there is a clinical
history of depression, convulsions, and other nervous signs that are exacerbated by
a high-protein diet, and may be alleviated by dietary control. Because there is no
cause of portal hypertension, these dogs do not develop ascites.
The liver that has been bypassed by a congenital shunt is hypoplastic, largely because
of diversion of hepatotrophic factors, including insulin, glucagon, and epidermal
growth factor that originate in the intestine and pancreas. Affected livers may be
smooth surfaced with normal color and texture. Histologically, hepatocytes and hepatic
lobules are small with close and irregular spacing of portal triads. Larger portal
veins may be inapparent or appear collapsed and empty of circulating blood elements;
portal veins in smaller triads may be small, collapsed, absent, or indistinguishable.
Hepatic arterioles are often more prominent, and may be multiple and tortuous (Fig.
2-13
), related to increased compensatory arterial perfusion. Numbers of arteriolar structures
within triads may also appear increased, as small caliber and usually inapparent arterioles
become evident histologically after compensatory hypertrophy. A proliferation of small
caliber bile ducts (ductular reaction) has been confirmed in some cases by cytokeratin
19 immunohistochemistry. Dilated vascular structures devoid of blood, presumably small-
and large-caliber lymphatics, are often prominent in the periphery of some portal
triads. Dilated lymphatics may also be present in the connective tissue surrounding
hepatic veins. In dogs, the spiral smooth muscle in the wall of the hepatic vein may
be more prominent in dogs with shunts than normal dogs. There may be increased deposition
of fibrous connective tissue surrounding portal triads and hepatic veins. Hepatocytes
may contain cytoplasmic lipid droplets, and multiple small lipogranulomatous foci
with hemosiderin and ceroid in Kupffer cells, and macrophages are typically present
throughout the liver, especially in animals >1 year of age.
Figure 2-13
Histology of the liver of a dog with a congenital portocaval shunt. Closely spaced
portal triads contain multiple sections of hepatic arterioles and lack discernable
portal veins.
Primary portal vein hypoplasia (PVH) has been reported in dogs, particularly Cairn
and Yorkshire Terriers, and occasionally in cats, affecting either the extrahepatic
or intrahepatic portal vein, or both. Intrahepatic portal vein hypoplasia is considered
to be the underlying lesion in conditions previously described as microvascular dysplasia,
hepatoportal fibrosis, and idiopathic noncirrhotic portal hypertension in some young
dogs. Depending on the level of the abnormality and extent of involvement of the lobes
of liver, PVH may be accompanied by portal hypertension, ascites and the development
of multiple collateral portosystemic shunts. Histologically, there is hypoplasia or
absence of portal vein radicles, secondary arteriolar proliferation, and atrophy of
hepatocytes. Moderate to marked portal fibrosis may also be present, with biliary
hyperplasia. These changes represent stereotypic sequelae to under-perfusion and thus
can be indistinguishable histologically from congenital portosystemic shunts; however,
development of portal hypertension is a distinguishing feature of PVH.
Macroscopic PSS and microscopic portosystemic vascular anomalies may co-occur in dogs,
as evidenced by a lack of resolution of clinical signs, and persistence of histologic
changes in additional liver biopsies subsequent to macroscopic shunt ligation. Decreased
tolerance of complete surgical shunt attenuation has been associated with lack of
identifiable portal veins and the presence of a ductular reaction in biopsies taken
during the initial surgical shunt attenuation procedure, although an earlier study
showed no association of severity of several histologic findings, such as arteriolar
proliferation, biliary hyperplasia, and fibrosis, with survival time after shunt attenuation.
Further reading
Awasthi A, et al. Morphological and immunohistochemical analysis of ductal plate malformation:
correlation with fetal liver. Histopathol 2004;45:260-267.
Baade S, et al. Histopathological and immunohistochemical investigations of hepatic
lesions associated with congenital portosystemic shunt in dogs. J Comp Pathol 2006;134:80-90.
Berent AC, Tobias KM. Portosystemic vascular anomalies. Vet Clin North Am Small Anim
Pract 2009;39:513-541.
Bertonlini G, Caldin M. Computed tomography findings in portal vein aneurysm of dogs.
Vet J 2012;193:475-480.
Best EJ, et al. Suspected choledochal cyst in a domestic shorthair cat. J Feline Med
Surg 2010;12:814-817.
Bosje JT, et al. Polycystic kidney and liver disease in cats. Vet Q 1998;20:136-140.
Bourque AC, et al. Congenital hepatic fibrosis in calves. Can Vet J 2001;42:145-146.
Brown DL, et al. Mesenchymal hamartoma of the liver in a late-term equine fetus. Vet
Pathol 2007;44:100-102.
Brown DL, et al. Congenital hepatic fibrosis in 5 dogs. Vet Pathol 2010;47:102-107.
Buczinski S, et al. Portacaval shunt in a calf: clinical, pathologic, and ultrasonographic
findings. Can Vet J 2007;48:407-410.
Bunch SE, et al. Idiopathic noncirrhotic portal hypertension in dogs: 33 cases (1982-1998).
J Am Vet Med Assoc 2001;218:392-399.
Center SA, et al. Hepatoportal microvascular dysplasia. In: Bonagura J, editor. Kirk's
Current Veterinary Therapy: XIII. Small Animal Practice. Philadelphia: WB Saunders;
2000. p. 682-686.
Christiansen JS, et al. Hepatic microvascular dysplasia in dogs: a retrospective study
of 24 cases (1987-1995). J Am Anim Hosp Assoc 2000;36:385-389.
Cullen JM. Summary of the World Small Animal Veterinary Association standardization
committee guide to classification of liver disease in dogs and cats. Vet Clin North
Am Small Anim Pract 2009;39:395-418.
Cullen JM, et al. Morphological classification of circulatory disorders of the canine
and feline liver. In: Rothuizen J, et al., editors. WSAVA Standards for Clinical and
Histological Diagnosis of Canine and Feline Liver Diseases. Philadelphia: Saunders
Elsevier; 2006. p. 41-59.
DeMarco J, et al. A syndrome resembling idiopathic noncirrhotic portal hypertension
in 4 young Doberman Pinschers. J Vet Intern Med 1998;12:147-156.
Desmet VJ. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc 1998;73:80-89.
Görlinger S, et al. Congenital dilatation of the bile ducts (Caroli's disease) in
young dogs. J Vet Intern Med 2003;17:28-32.
Grand JG, et al. Cyst of the common bile duct in a cat. Aust Vet J 2010;88:268-271.
Gunay-Aygun M. Liver and kidney disease in ciliopathies. Am J Med Genet Part C Semin
Med Genet 2009;151C:296-306.
Haechler S, et al. Congenital hepatic fibrosis and cystic bile duct formation in Swiss
Freiberger horses. Vet Pathol 2000;37:669-671.
Harper P, et al. Congenital biliary atresia and jaundice in lambs and calves. Aust
Vet J 1990;67:18-22.
Hunt GB. Effect of breed on anatomy of portosystemic shunts resulting from congenital
diseases in dogs and cats: a review of 242 cases. Aust Vet J 2004;82:746-749.
Hunt GB, et al. Evaluation of hepatic steatosis in dogs with congenital portosystemic
shunts using oil red O staining. Vet Pathol 2013;50:1109-1115.
Isobe K, et al. Histopathological characteristics of hepatic lipogranulomas with portosystemic
shunt in dogs. J Vet Med Sci 2008;70:133-138.
Kamishina H, et al. Gallbladder agenesis in a Chihuahua. J Vet Med Sci 2010;72:959-962.
Komine M, et al. Multiple hepatic peribiliary cysts in a young pig. Vet Pathol 2007;44:707-709.
Lamb CR, White RN. Morphology of congenital intrahepatic portacaval shunts in dogs
and cats. Vet Rec 1998;142:55-60.
Last RD, et al. Congenital dilatation of the large and segmental intrahepatic bile
ducts (Caroli's disease) in two Golden retriever littermates. J S Afr Vet Assoc 2006;77:210-214.
Lee KCL, et al. Association between hepatic histopathologic lesions and clinical findings
in dogs undergoing surgical attenuation of a congenital portosystemic shunt: 38 cases
(2000-2004). J Am Vet Med Assoc 2011;239:638-645.
Lyons LA, et al. Feline polycystic kidney disease mutation identified in PKD1. J Am
Soc Nephrol 2004;15:2548-2555.
McAloose D, et al. Polycystic kidney and liver disease in two related West Highland
white terrier litters. Vet Pathol 1998;35:77-81.
Mochizuki S, Makita T. Double gallbladder of swine. Kaibogaku Zasshi 1996;71:650-655.
Moore PF, Whiting PG. Hepatic lesions associated with intrahepatic arterioportal fistulae
in dogs. Vet Pathol 1986;23:57-62.
Parker JS, et al. Histologic examination of hepatic biopsy samples as a prognostic
indicator in dogs undergoing surgical correction of congenital portosystemic shunts:
64 cases (1997-2005). J Am Vet Med Assoc 2008;232:1511-1514.
Payne JY, et al. The anatomy and embryology of portosystemic shunts in dogs and cats.
Semin Vet Med Surg 1990;5:76-82.
Schermerhorn T, et al. Characterization of a hepatoportal microvascular dysplasia
in a kindred of Cairn terriers. J Vet Intern Med 1996;10:219-230.
Tisdall PLC, et al. Congenital portosystemic shunts in Maltese and Australian cattle
dogs. Aust Vet J 1994;71:174-178.
van den Ingh TSGAM, et al. Congenital portosystemic shunts in three pigs and one calf.
Vet Pathol 1990;27:56-58.
van den Ingh TSGAM, et al. Circulatory disorders of the liver in dogs and cats. Vet
Q 1995;17:70-76.
van den Ingh TSGAM, et al. Portal hypertension associated with primary hypoplasia
of the hepatic portal vein in dogs. Vet Rec 1995;137:424-427.
van den Ingh TSGA, et al. Morphological classification of biliary disorders of the
canine and feline liver. In: Rothuizen J, et al., editors. WSAVA Standards for Clinical
and Histological Diagnosis of Canine and Feline Liver Diseases. Philadelphia: Saunders
Elsevier; 2006. p. 61-76.
Yoshikawa H, et al. Congenital hepatic fibrosis in a newborn calf. Vet Pathol 2002;39:143-145.
Zandvliet MM, et al. Acquired portosystemic shunting in 2 cats secondary to congenital
hepatic fibrosis. J Vet Intern Med 2005;19:765-767.
Displacement, Torsion, and Rupture
The position of the liver should be observed as soon as the abdomen is opened at postmortem
examination. Caudal displacements resulting in extension of the margins of the liver
beyond the costal arch may be the result of hepatic enlargement or of displacement
of the diaphragm secondary to pleural effusion or other space-occupying lesions in
the thorax. Congenital or acquired displacements associated with ventral and diaphragmatic
hernias are common. Individual lobes or the entire organ may be displaced into the
subcutis, pleural cavity, or pericardial sac, often along with other viscera; lobar
blood supply may not always be compromised; however, individual displaced lobes may
be severely congested and may rupture, or, given time, become indurated.
Partial or complete liver lobe torsions have been reported in pigs, dogs, cats, and
horses. The left lateral lobe may be predisposed because of its mobility, large size,
and relative separation from other lobes; however, torsions of other lobes, in particular
the left medial lobe, as well as double-lobe torsions have been reported in dogs and
horses. Other predisposing causes include absence of or damage to the ligamentous
attachments that provide spatial support for the liver, trauma, or the presence of
a mass lesion in the affected lobe. Torsed lobes undergo various degrees of ischemia,
culminating in infarction caused by venous occlusion or venous and/or arterial thrombosis,
and affected animals may die because of shock, hemorrhage, or development of septic
peritonitis. Ischemia may favor overgrowth of Clostridium spp. with development of
necrosis and emphysema. Subacute cases may develop hepatic abscessation, and if the
animal survives, fibrosis and chronic inflammation.
Rupture
of the liver occurs commonly as the result of trauma because the organ is fragile
relative to its mass. Fatal liver rupture may be produced by the sudden accelerations
and pressures of vehicle collisions without much evidence of trauma to other parts
of the body. Large tears may be obvious in the liver capsule and hepatic parenchyma
after trauma; however, anastomosing linear patterns of fine shallow capsular fissures
may be concealed in part by clotted blood. Liver rupture is often clinically occult,
because quite large ruptures may not disturb liver function unless severe enough to
cause rapid exsanguination, or unless the biliary tract is involved. Intrahepatic
bile duct rupture results in bile extravasation into the hepatic parenchyma or beneath
the hepatic capsule, forming bile lakes or bile infarcts, areas of hepatocyte degeneration,
and necrosis surrounded by reactive macrophages; larger accumulations of bile may
be walled off by a pseudocapsule, forming biliary pseudocysts. Rupture of major bile
ducts or the gallbladder results in yellow-stained bile peritonitis, which may remain
sterile and become chronic, or may be fatal, particularly if infected by enterohepatic
circulation of bacteria such as clostridia.
The liver is more likely to rupture after trauma in young animals. Fatal ruptures
occur in foals during parturition, sometimes concurrently with costal fractures, and
in the smaller species subject to energetic emergency resuscitation. Diffuse hepatic
conditions with enlarged friable parenchyma (e.g., acute hepatitis, amyloidosis, severe
congestion, severe lipidosis, and infiltrating neoplasms) are more likely to rupture,
sometimes spontaneously, and the clinical consequences are related to the extent of
hemorrhage. Parasites that penetrate the capsule cause numerous small ruptures but
seldom lead to significant hemorrhage.
Further reading
Banz AC, Gottfried SD. Peritoneopericardial diaphragmatic hernia: a retrospective
study of 31 cats and eight dogs. J Am Anim Hosp Assoc 2010;46:398-404.
Bentz KJ, et al. Hepatic lobe torsion in a horse. Can Vet J 2009;50:283-286.
Bhandal J, et al. Spontaneous left medial liver lobe torsion and left lateral lobe
infarction in a Rottweiler. Can Vet J 2008;49:1002-1004.
Boerboom D, et al. Duodenal obstruction caused by malposition of the gallbladder in
a heifer. J Am Vet Med Assoc 2003;223:1475-1477.
Downs MO, et al. Liver lobe torsion and liver abscess in a dog. J Am Vet Med Assoc
1998;212:678-680.
Hinkle SG, et al. Liver lobe torsion in dogs: 13 cases (1995-2004). J Am Vet Med Assoc
2006;228:242-247.
Scheck MG. Liver lobe torsion in a dog. Can Vet J 2007;48:423-425.
Swann HM. Hepatic lobe torsion in 3 dogs and a cat. Vet Surg 2001;30:482-486.
Tennent-Brown BS, et al. Liver lobe torsion in six horses. J Am Vet Med Assoc 2012;241:615-620.
von Pfeil DJF, et al. Left lateral and left middle liver lobe torsion in a Saint Bernard
puppy. J Am Anim Hosp Assoc 2006;42:381-385.
Hepatocellular Adaptations and Intracellular Accumulation
The liver must be highly adaptable to balance function with changing demand. Increases
in the size of hepatocytes (hypertrophy) and their numbers (hyperplasia) collectively
bring a larger mass of hepatocytes into service. Such adaptations in hepatic volume
and function result from alterations in the expression of many genes. These responses
are more evident in smaller species, notably in laboratory rodents that have a very
pronounced liver growth response after exposure to various xenobiotics. The liver
can also adapt to reduced demand or oxygen supply by a combination of cellular atrophy
and apoptosis. Hepatocytes can be lost by apoptosis in substantial numbers, with minimal
elevation in serum enzymes of hepatic origin such as alanine aminotransferase.
Hepatocellular atrophy
Hepatic mass readily adapts to metabolic demands, and the liver can undergo marked
atrophy during illness and/or starvation without much evidence of impaired hepatic
function. During prolonged starvation, some hepatocytes are removed by apoptosis without
replacement, but most of the atrophy is explained by loss of cytoplasmic mass. Atrophic
livers, as seen, for example, in old grazing herbivores with poor teeth, are dark
and small, and the capsule may appear too large for the organ, showing fine wrinkles
on handling. These livers may even appear to be firmer than normal because of condensation
of normal stroma. Histologically, portal triads and hepatic venules are closer together,
and lobules contain increased numbers of smaller hepatocytes with scanty cytoplasm
(Fig. 2-14
). Hepatocellular mass can be rapidly lost by autophagy and apoptosis (see later section
on Types and patterns of cell death in the liver).
Figure 2-14
Hepatocellular atrophy in a dog with chronic right-sided heart failure.
Hepatic atrophy, rather than hepatocellular atrophy, can also result from impaired
replication of hepatocytes. Most adult hepatocytes are replicatively competent, although
mitotic figures are infrequent because healthy hepatocytes have a relatively long
life-span of several months. Diminished portal blood flow not only limits oxygen,
but also trophic factors that act to regulate replication and mass of the liver. These
trophic factors include several polypeptide growth factors, including hepatocyte growth
factor and insulin-like growth factors, and many hormones, including insulin, glucagon,
and catecholamines.
Atrophy of only a part of the liver may be a response to pressure or to impairment
of blood or bile flow. The histologic features of this atrophy are similar to those
of starvation atrophy. However, the functional consequences of focal hepatic atrophy
are minor because the remaining liver can compensate and adapt by a process that involves
replication and enlargement of hepatocytes. Local pressure atrophy occurs adjacent
to space-occupying lesions in the liver, or as a result of chronic pressures from
neighboring organs, such as distended rumen in the ox. It has been suggested that
right hepatic lobe atrophy, reported in horses, results from long-term compression
from abnormal distension of the right dorsal colon and base of the cecum (eFig. 2-3).
Chronic diffuse diseases of the biliary tract, such as sporidesmin poisoning and fascioliasis,
are likely to cause atrophy of the left lobe in ruminants, possibly as a result of
the greater difficulty in maintaining adequate biliary drainage from this lobe, whose
bile ducts are longer than those of the right in these species. The atrophy of biliary
obstruction is complicated by some degree of superimposed inflammation and fibrosis.
eFigure 2-3
Atrophic right lobe of the liver of a horse.
(Courtesy A.P. Loretti.)
Hepatocellular hypertrophy
Hypertrophy is the term used for the increase in liver size caused by an increase
in hepatocyte volume that may result from expansion of one or more organellar components
of the hepatocytes. Exposure to various xenobiotics can induce the expression of many
genes, leading to expansion of the smooth endoplasmic reticulum (SER), resulting in
hepatocyte hypertrophy. Agents that elicit this response act via nuclear receptors,
such as the arylhydrocarbon-activated receptor, the constitutive androstane receptor,
or the pregnane X receptor. Phenobarbital, for example, is a potent inducer of the
various enzyme systems of the SER, including several cytochromes P450 (CYPs). Hypertrophy
may occur in defined lobular regions, typically the centrilobular region or may affect
the entire lobule in more advanced cases, depending upon the activity and dose level
of the xenobiotic (Fig. 2-15
). Even when it is restricted to the centrilobular region, hypertrophy usually enlarges
the entire liver. Although hepatocellular hypertrophy is most often associated with
preferential increase in SER, proliferation of peroxisomes, or mitochondria can also
cause hepatocellular hypertrophy. The light microscopic appearance of hypertrophy
upon routine hematoxylin and eosin (H&E) staining will sometimes suggest the selective
involvement of one organelle. If total SER volume is increased, the cytoplasm will
typically have an eosinophilic ground-glass appearance upon light microscopy. If total
peroxisomal volume is increased, the cytoplasm is often noted to have an eosinophilic
granular appearance. The response can be seen within a few days after exposure to
various drugs and other xenobiotic compounds. Accordingly, the liver becomes grossly
enlarged. This induction of SER or peroxisomes is reversible, and after discontinuation
of exposure to the inducing agent, the expanded SER or excess peroxisomes are removed
by autophagy, and many hepatocytes undergo apoptosis. Although these changes are considered
physiologic adaptations, there are potential adverse sequelae; accordingly, this response
has toxicologic significance, and will be dealt with later in the section on Toxic
hepatic disease.
Figure 2-15
Increased cytoplasmic volume resulting from smooth endoplasmic reticulum induction
in a dog treated chronically with phenobarbital.
Polyploidy and multinucleation
Most mature mammalian hepatocytes are tetraploid or octaploid, whereas many immature
and replicating hepatocytes are diploid. The relative proportion of polyploid hepatocytes
varies among species, and polyploidy increases with age. Polyploidy is more common
in rodents and is believed to result from asynchrony of cell division in which binucleated
diploid cells undergo a second round of DNA replication, giving rise to 2 tetraploid
daughter cells. Impaired replication can also increase the number of polyploid cells.
The term megalocytosis was first used to describe the changes of liver cell cytoplasm
and nucleus that occur in pyrrolizidine alkaloid poisoning. This form of megalocytosis
has some specific features and is described in the later section on Chronic hepatotoxicity.
Impaired regeneration, atrophy and >4N polyploidy can also be produced by other DNA-damaging
agents such as aflatoxins. A feature of these patterns of atrophy with polyploidy
is the persistence of larger replicatively impaired polyploid hepatocytes amid regenerating
smaller diploid hepatocytes, hepatocellular nodules, and hyperplastic bile ductules.
Most hepatocytes are mononuclear, but a variable proportion is binucleated, especially
in young or regenerating livers of small animals. Multinucleation by more than 2 nuclei
of non-neoplastic hepatocytes is a rare phenomenon in domestic mammals, and its diagnostic
or pathogenetic significance is usually unclear. Multinucleation can result from incomplete
cell division or cell fusion, but this distinction is difficult to determine. Hepatocytes
can fuse during severe steatosis, but the degree of multinucleation in fatty livers
is hard to discern because the plasma membrane perimeters of fatty hepatocytes are
ill defined. Syncytial multinucleation of hepatocytes has been observed in various
degenerative and regenerative conditions. In protoporphyria of Limousin cattle, small
clusters of hepatocytes contain 4-10 or more closely packed nuclei, but it is not
clear whether this represents fusion or multiple nuclear divisions. Syncytial hepatocytes
are a characteristic of postinfantile giant cell hepatitis of children, and similar
hepatocyte multinucleation can be observed in association with some forms of hepatitis
in newborn cats, foals, and piglets (see Inflammatory diseases of the liver and biliary
tract). Multinucleated hepatocytes have been described in young cats with thymic lymphomas,
and in cats with experimental dioxin poisoning.
Intranuclear inclusions
In addition to the various nuclear inclusions associated with some viral infections,
3 types of inclusions may be found in hepatocyte nuclei.
1.
Spherical, apparently hollow globules within the body of the nucleus are membrane-bound
entrapped nuclear membrane invaginations that ultrastructurally contain cytoplasmic
components such as glycogen and mitochondria. These inclusions are infrequent in otherwise
normal livers but are more often seen in chronically injured livers, especially in
chronic pyrrolizidine alkaloid poisoning, in which polyploid nuclei are more likely
to indent and invaginate.
2.
Eosinophilic block-like intranuclear inclusions with a regular crystal lattice (“brick
inclusions”) are common in hepatocytes and renal proximal tubular epithelium. They
are more numerous in old animals, particularly dogs. Their composition and pathogenesis
are still unknown, but they evidently have little effect on the health of the cells
in which they occur, even when they are large enough to distort the nucleus. They
do not contain heavy metals and can be distinguished from the acid-fast, noncrystalline
intranuclear inclusions observed in renal epithelial cells and occasionally in hepatocytes
in lead poisoning.
3.
Lead inclusions consist of a lead-protein complex and have a characteristic furry
electron-dense ultrastructure.
Pigmentation
Congenital melanosis occurs in calves and occasionally in lambs and swine. The melanin
deposits may be numerous, and vary in size from flecks, to irregular, blue-black areas
2 cm or more in diameter. The melanin is confined to the capsule and the stroma. These
deposits are sharply defined in young animals, but become more diffuse and fade with
age (eFig. 2-4).
eFigure 2-4
Congenital melanosis in a calf.
A condition known as acquired melanosis is the massive accumulation of black pigment
(not melanin) in hepatocytes and Kupffer cells of mature sheep and, less frequently
cattle, after prolonged grazing on extensive unimproved pastures in inland eastern
Australia, the Falkland Islands, and Scandinavia. The condition in Norway has also
been described as hepatic lipofuscinosis. The color of the affected livers ranges
from a dull gray to uniform black, and there is usually a prominent acinar pattern.
In severe cases, there is also pigmentation of the hepatic lymph nodes, lungs, and
renal cortex. Histologically, the pigment is present as granules in lysosomes in periportal
and midzonal hepatocytes and macrophages of the liver, the proximal tubular epithelium
of the kidneys, and in alveolar and interstitial macrophages in the lung. There is
no evidence of liver dysfunction, even in the blackest livers. The source of this
pigment is not known, but the epidemiologic features of its occurrence indicate that
it is derived from a component of the diet that is sequestered within lysosomes.
Bile pigmentation may impart an olive-green color to the liver in diffuse or segmental
obstructive biliary disease or intrahepatic cholestasis. Histologically, conjugated
bile pigments may distend bile canaliculi, visible microscopically as golden-brown
linear streaks arrayed in a chicken wire-like pattern between the hepatocytes (Fig.
2-16
). In this case, the identity of the pigment is obvious, but when it is present in
granular form in Kupffer cell cytoplasm, it may easily be confused with hemosiderin
and hematin. Bile pigment is encountered infrequently in hepatocyte cytoplasm, almost
never in dogs, except in rare cases with dramatic cholestasis. Death of individual
hepatocytes releases the canalicular plugs into the space of Disse and the sinusoids,
where they may be phagocytosed by Kupffer cells.
Figure 2-16
Bile plugs distend bile canaliculi in a horse with intravascular hemolysis following
ingestion of red maple leaves.
Lipofuscin
is the term given to small, golden, granular cytoplasmic deposits derived from the
lipid component of membranous organelles, more obvious in hepatocytes near the centrilobular
areas. Lipofuscin accumulates in hepatocellular lysosomes and indicates senility,
atrophy, or increased turnover of membrane lipids. The pigment is particularly common
in the centrilobular regions of the liver of cats after they reach maturity. Ceroid
is a yellow pigment similar to lipofuscin and is associated with peroxidation of fat
deposits.
Black pigment also accumulates in hepatocellular lysosomes of mutant Corriedale sheep
with hyperbilirubinemia. These sheep have a condition resembling the human Dubin-Johnson
syndrome, in which there is mutation in the canalicular transporter in the organic
anion-transporting polypeptide family. This suggests that retention of the pigment
might be a sequel to a hepatic excretory defect.
In congenital erythrocytic protoporphyria (ferrochelatase deficiency) of Limousin
and Blonde d'Aquitaine cattle, a dark golden-brown lipofuscin-like lysosomal material
is present in portal areas, Kupffer cells, and sinusoidal endothelial cells, and is
heavily concentrated in the cytoplasm of hepatocytes.
Acquired protoporphyria (Fig. 2-17A, B
) with liver injury has been described in German Shepherd dogs as an idiopathic syndrome
and in a group of Beagle dogs treated with an experimental drug. Grossly, affected
German Shepherd dogs had dark livers that contained multiple regenerative nodules.
Histologically, there was abundant bridging portal fibrosis and typical orange birefringent
protoporphyrin crystals in hepatocytes. Photosensitization was not evident. Hepatic
vacuolation and nodule formation has also been described in a cat with porphyria.
Figure 2-17
A. Dark golden-brown protoporphyrin crystals in the liver of a dog. B. Protoporphyrin
crystals with bright red Maltese cross birefringence when viewed under polarized light.
Hemosiderin deposits are seldom sufficient to give gross discoloration, but when this
occurs, the color is dark brown. The pigment is detected microscopically as yellow
or brown crystals chiefly in the Kupffer cells, although small amounts may be found
in hepatic cells. The ferric iron component of this pigment can be demonstrated by
staining with Prussian blue; otherwise, it can easily be confused with lipofuscin.
Most hemosiderin deposits are punctate Prussian blue staining aggregates in Kupffer
cells but can also be seen as finer particles in hepatocytes. Diffuse hemosiderin
deposits in Kupffer cells occur quite commonly in all species, and its presence is
usually suggestive of excess hemolytic activity relative to the rate of reutilization
of iron. Thus it is seen in hemolytic anemia, anemia of copper deficiency, in cachexia,
and after blood transfusions or iron injections. It may be seen in Kupffer cells of
the centrilobular zones in severe chronic passive congestion of the liver. Localized
hemosiderin deposition occurs in areas of hemorrhage. Hemosiderin is normally present
in the liver in the early neonatal period, when fetal hemoglobin is being replaced
by mature hemoglobin.
Hemosiderin should be distinguished from hematin, which is produced by the action
of formic acid on hemoglobin following a prolonged postmortem interval, and is usually
regarded as a histologic artifact. Hematin is also an iron-containing pigment, but
the iron is in the reduced ferrous state and does not stain with ferricyanide. It
takes the form of crystalline brown deposits that are birefringent under polarized
light, mainly within hemoglobin-rich areas such as blood vessels. Hematin is darker
than hemosiderin and occurs in irregular clumps, often extracellularly. Hematin may,
however, be found in Kupffer cells and macrophages in small amounts.
Hepatic iron overload has previously been divided into hemosiderosis when there is
only an excess accumulation of hepatocellular iron and hemochromatosis when the excess
iron storage has produced fibrosis and inflammation and hepatic injury, although this
terminology is not used consistently. Hemochromatosis, as an inherited disorder is
relatively common in humans with specific mutations. Iron overload is common in some
birds, such as mynahs, and in lemurs, but rare in domestic mammals. A form resembling
heritable hemochromatosis has been reported in Salers or Salers-cross cattle. Affected
animals develop a wasting disease at ~1-2 years of age, have a 30-100 fold increase
in liver iron content, with dark-brown discolored, firm livers, hemosiderin accumulation
in hepatocytes, and periportal and perivenular bridging fibrosis with nodular regeneration.
Hemosiderin also accumulates in Kupffer cells, lymph nodes, kidney, pancreas, spleen,
and other organs.
Iron overload from dietary excess has been observed in sheep and cattle exposed to
high levels of iron in pasture and water. The liver is enlarged and brown with diffuse
fine nodularity, and the hepatic and adjacent lymph nodes are also darkened. Large
amounts of iron are present in the hepatic parenchyma, the biliary epithelium, and
the cortex of lymph nodes, and lesser amounts are present in the broad fibrous septa.
The iron is stored predominantly in lysosomes. Brown discoloration of bone marrow
resembles the osseous pigmentation of porphyria. The pathogenesis of nutritional iron
overload is unknown. Iron overload has also been described in horses, with animals
displaying signs of liver failure and neurologic impairment. The microscopic lesions
in the livers of these animals are similar to those for other species.
Brown crystalline deposits of 2,8-dihydroxyadenine (2,8-DHA) have been described in
hepatocytes and in other tissues in slaughtered cattle with no evidence of other disease.
These accumulations are strongly birefringent under polarized light, and are seen
in the cytoplasm of hepatocytes and macrophages of hepatic lymph nodes and as extracellular
deposits in portal stroma and renal tubular lumens. Grossly, the portal stroma stands
out as a green network, and affected lymph nodes were enlarged, and the medullary
sinusoids were distended with green pasty material. The crystals were identified as
2,8-DHA by a panel of crystallographic methods and mass spectrometry. The pathogenesis
is unknown.
Pigments of parasitic origin are particularly associated with flukes. Heavy deposits
of black iron-porphyrin compound are formed around the cysts and migratory pathways
of Fascioloides magna. Lesser amounts of similar pigment are deposited in bile ducts
infested by Fasciola hepatica (see later section on Helminthic infections). The presence
of this pigment in the hilar nodes should suggest otherwise inapparent infestations
by flukes. In schistosomiasis, the liver may be gray because of the accumulation of
black pigment in Kupffer cells.
Vacuolation
The term “vacuolar hepatopathy”
has been used to denote multifocal or diffuse zonal hepatocellular cytoplasmic vacuolation
before a more specific term can be applied, for example, before contents of vacuoles
are identified. Specific forms of hepatocyte vacuolar change include hydropic swelling,
glycogenosis, and steatosis (lipidosis). Hydropic degeneration can only be appreciated
in carefully controlled experimental circumstances and is an early cytoplasmic ballooning
seen after various toxic and metabolic insults, hypoxia, and cholestasis. Water and
sodium ion influx expands membranous compartments of mitochondria, lysosomes, and
endoplasmic reticulum (ER). The term feathery degeneration is applied to the type
of hydropic change that occurs in hepatocytes in which there has been prolonged cholestasis.
The cells are swollen and vacuolated and crisscrossed by a fine protoplasmic network
that is brown with bile pigments (Fig. 2-18
). This change is uncommon in cats and dogs.
Figure 2-18
Feathery degeneration in a horse with cholestasis.
Hepatic glycogenosis or glucocorticoid hepatopathy, often referred to as steroid-induced
hepatopathy, is probably the most severe example of hepatocellular vacuolar change
in dogs. It involves glycogen accumulation induced by hyperadrenocorticism, because
of either functional adrenocortical or pituitary tumors, or to treatment with glucocorticoids.
The liver is enlarged and pale tan. Hepatocytes are swollen, and the cytoplasm appears
as fine, diaphanous strands that enclose multiple spaces with poorly demarcated edges.
Vacuolar change may range from mild to severe, with swelling of hepatocytes from 2-10
times normal size and displacement of the nucleus and organelles to the cellular periphery
(Fig. 2-19
). The zonal distribution may be very variable and may become diffuse in long-standing
cases or with higher doses. Single-cell drop-out, multifocal small aggregates of neutrophils
along sinusoids, and scattered small foci of extracellular hematopoiesis are also
commonly observed. The ill-defined vacuolar boundaries in steroid hepatopathy are
readily distinguishable from spherical lipid vacuoles in hepatic steatosis. The pathogenesis
of this condition is uncertain, because glycogen storage alone does not fully explain
the influx of fluid, and possible perturbations in hepatocellular ion channels or
aquaporins have not been assessed. The amount of glycogen remaining in affected cells
is widely variable, a function of the original glycogen concentration, recent catabolism,
and postmortem dissolution. Glycogen content can best be demonstrated in frozen section,
followed by staining with periodic acid–Schiff, and can be confirmed as glycogen by
its sensitivity to digestion with diastase. Livers affected by glucocorticoid hepatopathy
maintain normal hepatocyte functions, but the condition can be confirmed by the induction
and serum increase of glucocorticoid-inducible alkaline phosphatase, with minor or
negligible increases in alanine aminotransferase. Similar vacuoles may be created
by other adrenocortical steroids, possibly progestins, as occasionally seen in older,
female dogs that appear to have overproduction of adrenal cortical hormones other
than cortisol and alkaline phosphatase elevation, although the specific pathogenesis
is unknown. Some drugs, such as the chelator D-penicillamine, can produce similar
vacuolation.
Figure 2-19
Prominent vacuolation of hepatocytes following glucocorticoid administration in a
dog.
Loss-of-function mutations in hepatic and renal glucose-6-phosphatase resulting in
increased hepatic glycogen storage have been reported in Maltese puppies or related
crossbred dogs, and these dogs serve as an animal model of glycogen storage disease
(glycogenosis) type Ia. These dogs have severely debilitating problems in maintaining
their blood glucose because dephosphorylation of glucose-6-phosphate is a key step
in both glycogenolysis and gluconeogenesis. The condition is typified by severe hepatocellular
glycogenosis. Livers are markedly enlarged, pale, and have diffuse vacuolation of
hepatocytes with large amounts of glycogen and small amounts of lipid. Hepatic fibrosis
and nodular regeneration can develop with time. Renal tubular epithelium is also vacuolated.
Glycogen storage disease type III, with hepatic glycogen storage, has been reported
in dogs, predominantly German Shepherd dogs. Type IV glycogenosis has been reported
in Norwegian Forest cats, although this disorder causes pale blue granules in hepatocellular
cytoplasm rather than clear vacuoles.
Characteristic hepatocellular vacuolation is also described in dogs with end-stage
nodular livers presented clinically with hepatocutaneous syndrome.
Hepatocellular steatosis (lipidosis)
Hepatocellular steatosis
is the term used to describe fatty livers of animals. However, the terms steatosis,
lipidosis, and fatty change tend to also be used interchangeably. All of these terms
refer to the visible accumulation of triglycerides (triacylglycerols) as round globules
in the cytoplasm of hepatocytes. The threshold for application of these terms is vague
because triglyceride storage and transport are normal hepatic functions, but they
are appropriate when the amounts are greater than would normally be seen. Hepatocellular
steatosis can be physiologic or pathologic. Any circumstance in which hepatic uptake
of lipids exceeds oxidation or secretion can lead to hepatocellular steatosis. Increased
mobilization of triglycerides during late pregnancy or heavy lactation in ruminants
is associated with hepatocellular steatosis. The lipid represents increased transit
of triglycerides in an otherwise healthy liver, so there is little diagnostic significance
in mild degrees of hepatocellular steatosis in lactating cows. In some of these animals,
severe energy deficiency can also lead to clinical ketosis with metabolic acidosis.
In addition, severe steatosis occurs in high-producing dairy cows fed diets in which
either the mix of available fatty acids is incorrect or lipids are oxidized and rancid.
Hepatocellular steatosis is common in injured hepatocytes because the normal high
throughput of fatty acids and triglycerides can be readily impeded at various points
in the complex pathway of hepatic lipid metabolism and secretion of very-low-density
lipoproteins (VLDLs). Hepatocytes obtain some fatty acids from albumin and other carrier
proteins in the portal blood, but most is hydrolyzed by sinusoidal endothelial hepatic
lipase from triglyceride in chylomicrons or VLDL in plasma. Very-long-chain fatty
acids are initially oxidized by acyl–coenzyme A oxidase in peroxisomes to shorter
acyl–coenzyme A that can be transported via a carnitine-dependent process into mitochondria
for further oxidation. Although some fatty acid is used for energy in hepatic mitochondria,
most from dietary or adipose sources is converted to triglycerides that are further
processed into lipoproteins in the hepatocyte ER and actively secreted into the plasma
as VLDL. Endothelial lipoprotein lipase in muscle and adipose tissue hydrolyzes triglycerides
in the VLDL, and the fatty acids released are used locally by β-oxidation in mitochondria
of muscle or re-esterified into triglyceride in adipocytes. The depleted VLDLs are
returned by receptor-mediated endocytosis to hepatocytes, where their constituents
are catabolized and recycled.
The synthesis and export of VLDL in hepatocytes are energy- and resource-dependent,
so any disturbance of the supply of apoproteins (apoprotein B primarily), phospholipids,
cholesterol, or ATP, or physical disruption of the organelles involved in synthesis,
assembly, and secretion, has the potential to inhibit lipoprotein synthesis or secretion.
Fatty acids can continue to enter hepatocytes, allowing triglyceride globules to accumulate
in the hepatocyte cytoplasm. In some species, damage to peroxisomes reduces the initial
peroxisomal oxidation step for catabolism of long-chain fatty acids and also upregulates
various genes regulated by peroxisome proliferator–activated receptor-α. Lipoproteins
in the ER, Golgi, and membrane-bound secretory vesicles can also accumulate if there
is selective damage to the distal secretory apparatus. Damage by various toxic and
hypoxic insults will not lead to triglyceride accumulation in hepatocytes if supplies
of mobile triglycerides from adipose tissue or the diet are low.
The microscopic appearance of triglyceride globules in hepatocytes ranges from small
discrete microvesicles to large coalescing macrovesicles. Microvesicular lipid vacuoles
are smaller than the nucleus and tend not to displace the nucleus (Fig. 2-20
). Microvesicular steatosis can be a hallmark of more severe hepatic dysfunction than
macrovesicular steatosis. This pattern can occur in several toxic hepatopathies causing
mitochondrial injury, including some drug toxicities, such as antiviral nucleosides,
aspirin in Reye's syndrome, and excessive tetracycline administration, all of which
can be fatal. These toxicities tend to target: mitochondria, disrupting energy production;
the normal electron transfer chain, causing oxidative injury; and interfere with β-oxidation
of lipids. Partially oxidized lipids typically have a lower surface tension than triglycerides
and therefore form smaller vesicles in the aqueous medium of the cytoplasm. Uncontrolled
diabetes mellitus and feline fatty liver syndrome can produce microvesicular hepatic
steatosis or a mixture of microvesicular and macrovesicular steatosis. Acute steatosis
with predominantly microvesicular accumulation tends to result in a modestly enlarged
pale liver without much change in texture, whereas macrovesicular hepatic steatosis
more often produces an enlarged liver.
Figure 2-20
Microvesicular lipid is characterized by multiple round, clear vacuoles that are smaller
than the nucleus and do not displace the nucleus.
In some more protracted toxic injuries, smaller lipid globules can coalesce into large
central macrovesicles that displace the nucleus. The pathogenesis of the larger globules
is not well understood, but it involves some alteration of the globule-cytoplasm interface
that normally prevents coalescence of the small micellar globules, possibly the greater
hydrophobicity of the triglycerides. These fatty livers tend to be more yellow and
enlarged, and the texture is more friable than livers with microvesicular steatosis.
Each hepatocyte usually contains one large globule (macrovesicle) that alters the
contour of the cell and displaces the nucleus (Fig. 2-21
). The sinusoids are compressed and appear under-perfused, and the tissue at low magnification
resembles adipose tissue. In severe degeneration, the liver is moderately or greatly
enlarged, with a uniform light yellow color. The edges are rounded, and the surface
is smooth (eFig. 2-5). The cut surface has a diffuse greasy appearance or a red and
yellow lobular pattern if there is also hepatic congestion or zonal necrosis. In severe
diffuse hepatic steatosis, the parenchyma is less dense, and portions will float in
water or fixative.
Figure 2-21
Macrovesicular lipid vacuoles in the liver of a donkey. Vacuoles are larger than the
nucleus and tend to displace it to the periphery of the cell.
eFigure 2-5
Hepatomegaly, steatosis associated with equine hyperlipemia in a donkey.
Assessment of the functional significance of steatosis depends on the differentiation
of physiologic steatosis caused by increased mobilization by otherwise normal hepatocytes
from pathologic changes that represent some degenerative change in hepatocytes. However,
fat accumulation is a sensitive response to hepatocellular injury and can occur in
the absence of other obvious alterations in hepatic structure or function. The triglyceride
globules themselves are not harmful to hepatocytes, so the amount of fat present is
more an indicator of the duration of insult and triglyceride supply than of the severity
of hepatic injury. Steatosis is usually reversible, although a liver that has been
fatty for some time is more likely to have concurrent damage, including fibrosis,
pigment accumulation, and nodular hyperplasia, mostly attributable to ongoing peroxidative
damage, cell turnover, and activation of stellate cells.
Hepatocellular steatosis is now recognized as a more significant indicator of potential,
more severe liver injury than previously appreciated. In ruminants, the association
between hepatic steatosis, ketosis, and displaced abomasum are well recognized. Infectious
diseases, such as metritis and mastitis, may be more likely or prolonged. Neutrophil
function is suppressed, as is interferon production by lymphocytes, and endotoxin
clearance is also reduced. Hepatocytes become more sensitive to injury following exposure
to cytokines such as tumor necrosis factor-α or endotoxin as well. Consequently, fatty
livers are more vulnerable to a wide range of toxic and nutritional insults, so other
necrogenic insults and responses can be concurrent. Increased levels of fatty acids
in the liver increase the oxidative stress and contribute to membrane lipid peroxidation,
reduced hepatocyte life-span, and some local repair responses.
When lipid accumulates in large amounts, there is a tendency for groups of the fat-laden
cells to rupture or fuse and eventually form a multinucleated rim about a foamy mass
of lipid. This epithelial structure is known as a fatty cyst (Fig. 2-22
). The next stage, which occurs when released lipid is picked up by macrophages that
form foamy aggregations in sinusoids, in the stroma of portal tracts, and hepatic
venules is termed a lipogranuloma. Subsequent peroxidation of the less saturated fatty
acids and covalent modification and polymerization of oxidized lipids form a complex
of lysosomal residues known collectively as ceroid. This material is only slightly
soluble in lipid solvents and is periodic acid–Schiff positive, diastase resistant,
variably acid fast, and autofluorescent. In histologic sections, ceroid pigment appears
as colorless or yellow irregular fragments associated with the lipid globules in macrophages
and, to a lesser extent, hepatocytes. Affected portal lymph nodes become slightly
enlarged, yellow-green, and rather oily on section. If iron accumulates along with
the ceroid, the lesion is more correctly termed a pigment granuloma (Fig. 2-23
). These are discrete perisinusoidal clusters of macrophages, containing cytoplasmic
lipid vacuoles, lipofuscin, and hemosiderin, and often contain lymphocytes or plasma
cells. They accumulate and congregate with age and hepatocellular turnover, but have
no known clinical significance.
Figure 2-22
Fatty cyst in the liver of a dog.
Figure 2-23
Pigment granuloma (lipogranuloma) in the liver of a dog.
Some chronic changes commonly seen in the livers of old dogs can be associated with
fatty liver; these include lipogranulomas, fatty cysts, and ceroid accumulation. Occasionally,
there can be found sharply defined, fibrous, stony, hard masses, usually close to
the surface, sometimes as much as 3-4 cm in diameter. These masses are usually sufficiently
mineralized to show up distinctly on clinical radiographs. The mineral appears to
be deposited on a matrix of degenerate collagen, laid down about perivascular foci
of foamy macrophages and accumulations of cholesterol. The fibrous tissue may be laid
down in response to the continued presence of fat, ceroid, or cholesterol, but no
reason is apparent for the strictly localized distribution of the reaction. There
are no recognizable hepatocytes in these lesions.
Physiologic steatosis (fatty liver)
occurs in late pregnancy and heavy lactation, particularly in ruminants and llamas.
In this circumstance, the lipid is typically macrovesicular, with large, round, clear
vacuoles that tend to displace the nucleus. Obvious steatosis is also seen in neonates,
especially in those species whose milk is relatively rich in fat. These livers are
fatty enough to be pale to the naked eye. The high rate of mobilization of triglycerides
from body fat stores is mainly responsible for fatty liver in lactating ruminants.
However, fatty liver is a concern because lipid mobilization also increases when the
dietary energy intake is insufficient relative to the production demands that are
greatest in early lactation of high-producing cows. Insufficient dietary intake by
an animal with adequate fat reserves depletes hepatocellular glycogen and initiates
a heavy demand for triglycerides from adipose tissue. When hepatic triglyceride concentrations
exceed 10% on a wet weight basis, severe or clinical fatty liver ensues. At this time,
urinary ketones are elevated, and body weight loss and appetite depression can occur.
In severe cases, cattle can suffer from hepatoencephalopathy. It is estimated that
in the first month after parturition 5-10% of dairy cattle severe fatty liver, and
30-40% have moderate fatty liver (5-10% liver triglyceride by wet weight basis). The
liver depends primarily on fatty acid oxidation for its own considerable energy needs.
It must also synthesize a large amount of protein and phospholipids for lipoprotein
export to other tissues, and this can be rate limiting, resulting in accumulation
of triglyceride in the cytoplasm. In starvation, the reduced availability of protein
and lipotrope cofactors, such as choline, can exacerbate the bottleneck. The liver
is the main supplier of glucose for the brain and milk saccharides, and it adapts
by converting fatty acid metabolites to glucose (gluconeogenesis) and ketones.
Acute ketosis of lactating dairy cows with intake insufficiency or secondary to abomasal
displacement is usually associated with fatty liver with a predominantly diffuse microvesicular
pattern. Cows are more tolerant of ketosis associated with lactation than are pregnant
ewes that can die from starvation-induced
pregnancy toxemia
and ketoacidosis. In cows, hepatic steatosis is predominantly centrilobular, whereas
it is most severe in the periportal zone in pregnancy toxemia of sheep. The hepatic
changes reflect increased mobilization of triglycerides from adipose tissue rather
than hepatic disease per se. In cows and ewes with increased mobilization of triglycerides,
there may be indistinct foci of white discoloration of abdominal fat that tend to
be obscured when adipose tissue solidifies postmortem.
Fatty liver of diabetes occurs when insulin is deficient or inactive because of lack
of functioning receptors. Reduced insulin-dependent glucose uptake by cells leads
to accelerated lipolysis from adipose tissue in much the same way as when energy intake
is limiting. The liver is thus presented with a large load of fatty acids, and the
rate by which this moves through the liver can be impeded in various ways. Insulin
deficiency alone will produce fatty liver. Some cases in carnivores are also complicated
by concurrent exocrine pancreatic insufficiency, so protein malabsorption can be a
contributing influence on diabetic hepatic steatosis. The centrilobular hepatocytes
usually show the greatest degree of steatosis, but in advanced long-standing diabetes,
the change is often diffuse and marked.
Lipoprotein synthesis and transport are dependent on oxidative metabolism, so hypoxia
of hepatocytes leads to triglyceride accumulation. The 2 most common causes of hepatocellular
hypoxia are anemia and reduced sinusoidal perfusion in passive venous congestion.
In these situations, hepatic steatosis is most severe in the centrilobular zone, provided
that the adipose and dietary supply of triglyceride is sufficient. Hepatic steatosis
can be grossly visible as a yellow zonal pattern in chronic passive congestion of
the bovine liver, but is less obvious in carnivores with passive hepatic congestion.
Local hypoxia is probably the basis for another example of fatty liver. Small, sharply
demarcated patches of intense fatty infiltration are often seen in bovine livers at
or adjacent to sites of capsular fibrous adhesions—so-called “tension lipidosis.”
These patches are neither swollen nor shrunken, usually extend <1 cm into the parenchyma,
and are of the same consistency as normal liver (Fig. 2-24
). The acinar structure of these lesions is undisturbed, but the hepatocytes therein
show pronounced steatosis, presumably related to interference with local perfusion
caused by tensions transmitted to the parenchyma by the adhesion.
Figure 2-24
Subcapsular focal fatty change (“tension lipidosis”) associated with capsular ligamentous
attachment in an ox.
(Courtesy A.P. Loretti.)
Hepatic steatosis caused by intoxication is common. There are several stages of the
cycle of hepatic lipid metabolism that can be affected selectively by various toxins
to produce fatty liver. For example, it is possible experimentally to cause triglyceride
accumulation by interfering with mitochondrial fatty acid oxidation with sublethal
doses of cyanide, or by inhibiting apolipoprotein synthesis by administration of orotic
acid. Most toxins that cause fatty liver in naturally occurring situations, however,
also produce a greater or lesser degree of hepatocellular necrosis. Fatty liver occurring
as a manifestation of toxic hepatic disease will be further discussed in the later
section on Toxic hepatic disease, but the generalization may be made here that most
important veterinary hepatic intoxications cause widespread membrane damage and/or
disturbance of protein synthesis. These cause lipid accumulation in the hepatocyte
by interfering with lipoprotein synthesis and export, as well as with fatty acid oxidation.
Steatosis requires time and a negative energy balance to develop, so it is more likely
to occur in toxicoses with a longer clinical course. However, if adipose reserves
are depleted, there is less lipid available to accumulate in the liver.
Although fatty liver in domestic animals is more frequently associated with generalized
interferences with energy metabolism, there are some specific nutritional deficiencies
that will produce fatty liver. These have usually been defined under experimental
conditions. Choline deficiency, in conjunction with deficiency of other lipotropic
factors, such as L-methionine and vitamin B12
, rapidly produces fatty liver, largely as a result of reduced synthesis of phosphatidylcholine,
a component of secreted lipoproteins. Fatty liver in experimental choline deficiency
involves lipid peroxidation and increased hepatocellular turnover, leading to cirrhosis
and neoplasia. It is unlikely that primary choline deficiency occurs in domestic animals,
but other lipotrope deficiencies have been reported.
Ovine white-liver disease, first described in lambs in New Zealand, also occurs in
southern Australia, the United Kingdom, and continental Europe. Goats are also susceptible.
Ovine white-liver disease is characterized by a syndrome of ill-thrift to emaciation,
anorexia, and mild normocytic normochromic anemia, occasionally with photosensitization
and icterus. The condition is associated with low liver cobalt levels and low plasma
concentrations of vitamin B12, and the disease has been shown to be cobalt and vitamin
B12 responsive. Lambs up to 1 year of age are more commonly affected than ewes, and
pastures are likely to be adequate at the times of peak incidence in late spring and
early summer.
In the early stages, the liver changes consist of vacuolar accumulation of triglyceride
in hepatocytes, usually most severe in the centrilobular zones. In addition, ceroid
pigment is present in all cases, early in hepatocytes and later also in sinusoidal
cells and macrophages. The fatty change may be very severe in the early stage, the
liver being grossly swollen. A moderate degree of bile ductular proliferation is also
a consistent feature, and the epithelium of the smaller ductules in the triads is
dysplastic. Spongy degeneration of cerebral white matter, typical of the hyperammonemia
of hepatic failure, is present in some cases.
Experimental feeding of a diet low in cobalt to sheep resulted in reduced growth rate,
anorexia, lacrimation, alopecia, emaciation, and marked reduction in plasma and liver
vitamin B12 concentrations. At autopsy, livers were pale, swollen, and fatty. Histologically,
livers with severe fatty degeneration were characterized by widespread hepatocyte
disassociation, accumulation of lipid droplets, eosinophilic inclusions and lipofuscin
in hepatocyte and Kupffer cell cytoplasm, nuclear lipid inclusions, ductular proliferation,
and hepatocyte apoptosis. These lesions are characteristic of spontaneous cases of
ovine white-liver disease. Ultrastructurally, degeneration of mitochondria and proliferation
of the smooth endoplasmic reticulum are evident. The disease can be produced in cobalt-deficient
sheep fed diets high in propionate precursors, which may help explain the explosive
nature of outbreaks on lush pasture.
Hepatic lesions similar to lipotrope-deficient forms of experimental nutritional cirrhosis
have been reported in sheep, goats, cattle, deer, and pronghorn antelope from Texas,
New Mexico, and northeastern Mexico. Hard yellow-liver disease, or hepatic fatty cirrhosis,
is a progressive, chronic disease characterized by weight loss and hepatic encephalopathy
(eFig. 2-6). The disease typically appears in years following above-average winter
rains, followed by drought conditions in the summer months. Grossly visible liver
lesions in sheep begin in the subcapsular hepatic parenchyma along the porta hepatis
as pale yellow, firm areas, spreading peripherally to involve ~80% of the liver in
the final stages of the disease. Microscopic changes include accumulation of fine
cytoplasmic lipid droplets in centrilobular hepatocytes, later involving the entire
lobule, with rupture and formation of fatty cysts. Centrilobular fibrosis accompanies
the ruptured fatty cysts, progressing to widespread bridging centrilobular fibrosis,
with islands of regenerating hepatocytes. Kupffer cells and macrophages in regional
lymph nodes, spleen, and lung contain abundant ceroid. The etiology of this condition
is unknown, although unidentified hepatotoxins, possibly altering lipoprotein synthesis
and secretion, combined with nutritional stress, have been postulated.
eFigure 2-6
Hard yellow-liver disease in a goat.
(Courtesy P. Stromberg.)
Equine hyperlipemia
is almost exclusively a disease of donkeys, miniature horses, and ponies, and among
these, the Shetland breed predominates. The syndrome is characterized by marked elevation
of serum triglyceride concentration, predominantly very-low-density lipoprotein (VLDL),
but other lipid fractions are also elevated, with visible prominent lipemia and hepatic
steatosis. The condition is usually fatal after about a week. A negative energy balance
is a key feature. Pregnant or lactating mares are most likely to develop the disease,
particularly if they are older, excessively fat, and have recently suffered reduced
feed intake because of onset of parturition, conditions such as laminitis or parasitism,
or other causes of stress. The clinical course is marked by somnolence, complete anorexia,
and colic, progressing to mania in some cases, although most simply become progressively
more depressed. Some ponies develop ventral subcutaneous edema and most develop moderate
diarrhea. Metabolic acidosis is a consistent feature in animals that die.
The liver at autopsy is severely fatty and may have ruptured; the steatosis also extends
to heart and skeletal muscle, kidney, and adrenal cortex. The hepatic steatosis is
remarkable only by its severity; there may be some focal hepatocellular necrosis,
and there is consistent prolongation of bromosulfophthalein retention times and elevation
of serum alkaline phosphatase levels. Evidence of disseminated intravascular coagulation
is seen as serosal hemorrhages and microscopic thrombi in various organs, and even
gross infarction of myocardium and kidney. Small lipid emboli may be detected in frozen
sections of lung, myocardium, and brain in these animals; their relationship to the
microthrombosis is uncertain.
The pathogenesis of this disease is obscure. Because the excess lipid in liver and
blood is in the form of triglyceride, the implication is that the liver is capable
of esterifying fatty acid mobilized from depot fat. The triglyceride thus formed is
presumably then exported to the plasma as VLDL. It is believed that the primary cause
of hyperlipemia is increased production via an increased rate of adipocyte lipolysis,
leading to fatty acids and glycerol being released into the bloodstream and the increased
hepatic synthesis of triglyceride as VLDL, rather than reduced clearance of VLDL from
the serum. Another possibility is that there is an inability on the part of all tissues
other than the liver to use fatty acids from VLDL at the normal rate, whereas triglyceride
synthesis from fatty acids continues in the liver. In any event, at this stage lipids
begin to accumulate in hepatocytes.
It has been proposed that an underlying cause of pony hyperlipemia is a comparative
resistance to insulin in susceptible animals, and that this is compounded in stressful
episodes by increased levels of circulating cortisol. Various steroid hormones, including
glucocorticoids, have been shown to interfere with insulin action, and hyperlipemic
ponies often have elevated plasma insulin levels, which suggests reduced function
of insulin receptors. However, plasma ketones are much less consistently elevated,
which appears to be characteristic of equids; this suggests that the increased ketogenesis
that one might expect in insulin resistance is not a typical feature in horses.
Hepatic steatosis is common in companion animals. Following periods of stress or fasting,
toy-breed dogs can develop profound hypoglycemia and a striking microvesicular hepatic
steatosis. This may be the result of poor homeostasis of blood glucose levels. Most
often, affected pups die from cerebral complications of hypoglycemia, but significant
liver dysfunction also occurs. Dogs eating diets deficient in vitamin E may develop
severe hepatic steatosis, but there are many circumstances in companion animals where
the exact cause for individual cases of steatosis cannot be determined.
The syndrome of
feline hepatic steatosis
most commonly occurs in obese, nutritionally stressed female cats, which display vomiting,
anorexia, weakness and weight loss, jaundice, and hepatomegaly (Fig. 2-25
). Neurobehavioral signs indicative of hepatic encephalopathy, other than drooling
and depression, are rarely reported. Affected cats generally have hyperbilirubinemia,
and a significant increase in serum alkaline phosphatase activity, in the face of
normal or modest increases in γ-glutamyltranspeptidase activity, a key diagnostic
feature of this syndrome. Untreated, the mortality rate is high. The liver has diffuse,
macrovesicular or microvesicular steatosis, by definition affecting >50% of hepatocytes.
Focal or zonal lipid accumulation in <50% of the parenchyma is considered more likely
to be physiologic, or associated with other systemic abnormalities, rather than idiopathic
feline hepatic steatosis. Bile pigment accumulates in canaliculi or Kupffer cells
and can be confused with lipofuscin and ceroid.
Figure 2-25
Hepatomegaly resulting from hepatic steatosis in a cat
(Courtesy A.P. Loretti.)
The pathogenesis of hepatocellular triglyceride accumulation in this disease is obscure,
and likely multifactorial, involving increased mobilization and uptake of nonesterified
fatty acids by the liver, alterations in formation and release of VLDL, and impaired
oxidation of fatty acids within hepatocytes. Ultrastructural studies have demonstrated
decreased numbers and abnormal morphology of hepatic peroxisomes as well as mitochondria,
both of which are important in the oxidization of fatty acids, but whether these changes
are significant or simply adaptive responses is unknown. Starvation may reduce the
availability of proteins, choline, and other precursors necessary for lipoprotein
synthesis.
Severe hepatic steatosis can also develop in cats, concurrent with or secondary to
other major medical problems such as diabetes mellitus, which alter metabolism of
fat. Acute pancreatitis appears to be an important predisposing disease to secondary
hepatic steatosis in cats—an association with clinical significance as it has a poorer
prognosis than uncomplicated idiopathic hepatic steatosis. Secondary hepatic steatosis
has also been reported with concurrent inflammatory liver disease, such as cholangitis,
renal disease, small intestinal disease, neoplasia, and hyperthyroidism.
Familial hyperlipoproteinemia has been described in cats, associated with congenital
lipoprotein lipase deficiency. The condition is characterized by lipid vacuoles and
ceroid accumulation in liver, spleen, lymph nodes, kidney and adrenal glands, multifocal
xanthomas, and focal arterial degenerative changes. An autosomal recessive mode of
inheritance is suspected. Primary idiopathic hyperlipidemia has also been reported
in Miniature Schnauzer dogs and Beagles, although the metabolic defect has not been
identified. Affected dogs have fasting lipemia, elevated plasma VLDL, and may have
hyperchylomicronemia. Affected animals may develop severe vacuolar hepatopathy associated
with both glycogen and triglyceride accumulation, with eventual stromal collapse and
regenerative nodule formation. There is also an association between hyperlipidemia
and gallbladder mucocele.
An incompletely characterized condition known as hepatic lipodystrophy has been recognized
in pedigree Galloway calves since 1965. Calves initially appear normal but develop
lethargy, tremors, and opisthotonos, and die by 5 months of age. On postmortem examination,
affected calves have an enlarged, pale, mottled liver. Histologically, there is marked
hepatic steatosis with portal fibrosis and ductular reaction (Fig. 2-26
). Vacuolar changes in the white matter of the brain are consistent with hepatic encephalopathy.
A metabolic defect has been proposed.
Figure 2-26
Hepatic steatosis, portal fibrosis, and bile duct hyperplasia resulting from hepatic
lipodystrophy in a Galloway calf.
(Courtesy M.J. Hazlett.)
Lysosomal storage diseases
In common with other tissues in animals with a heritable deficiency of specific lysosomal
enzymes, liver cells may accumulate substrates normally catabolized by the missing
enzyme. These lysosomal stores can be less obvious in the liver than in other tissues
such as the central nervous system, and although they are unlikely to affect hepatic
function, they can sometimes be recognized in liver biopsies (eFig. 2-7). However,
hydropic and fatty changes in hepatocytes can obscure or lead to misidentification
of lysosomal storage vacuoles. Kupffer cells and bile duct epithelium may be more
severely affected than hepatocytes, which have additional catabolic and excretory
pathways, including the ability for lysosomal exocytosis into the bile canaliculi.
In animals with ceroid lipofuscinosis, lysosomal storage is minimal in hepatocytes
compared to that in the brain. Examples of storage disorders associated with hepatomegaly
and abnormal hepatic or Kupffer cell lysosomal inclusions include GM1 gangliosidosis
(β-1-galactosidase deficiency) in cats, mucopolysaccharidosis type I (α-L-iduronidase
deficiency) in dogs, and α-mannosidosis in cats. These and others are discussed in
more detail in Vol. 1, Nervous system.
eFigure 2-7
Vacuolated hepatocytes in a cat with a ganglioside lysosomal storage disease (GM2),
causing abnormal lysosomal accumulation.
Hepatic phospholipidosis is an excessive accumulation of phospholipids within the
cytoplasm of hepatocytes. This condition is an acquired storage disorder caused by
some therapeutic drugs and chemicals, including amiodarone and chlorphentermine. Despite
the diversity of agents capable of causing phospholipidosis, most of these are cationic
amphipathic molecules. Mechanistically, phospholipidosis is caused by cationic amphipathic
molecules binding to cellular phospholipids and inhibiting complete digestion by lysosomal
phospholipase A1, A2, or C within lysosomes; although less often, direct inhibition
of phospholipase can occur, as in the case of gentamicin. This leads to an accumulation
of lysosomal phospholipids that can develop acutely or only following long-term drug
administration. The histologic appearance of phospholipidosis can be quite variable,
but typically, hepatocellular and Kupffer cell cytoplasm contains numerous round clear
vacuoles that are smaller than the diameter of the nucleus or larger, imparting a
foamy appearance to the cytoplasm. Fine vacuoles are most often apparent adjacent
to canaliculi. Zonal distribution can vary depending on the type of drug or chemical.
Biliary epithelium can also be affected, with or without hepatocellular involvement.
The clear vacuoles can be confused with microvesicular steatosis, and in most cases,
ultrastructural examination is needed to identify phospholipidosis with certainty.
At the ultrastructural level, there is a characteristic multilaminated whorl of material
with a “fingerprint” pattern, termed myeloid bodies, within affected lysosomes.
Amyloidosis
In most species, hepatic amyloidosis is usually part of generalized amyloidosis.
Systemic amyloidosis of domestic animals is typically associated with overproduction
of amyloid A (AA), an amino-terminal fragment of serum amyloid A, a highly inducible
acute-phase protein in most species. In humans, AA amyloidosis occurs either as a
familial trait (familial Mediterranean fever), or secondary to a sustained acute-phase
reaction in chronic inflammatory or neoplastic diseases. Amyloid infiltration of the
liver occurs sporadically in cattle, horses, dogs, and cats as a secondary response
to chronic disease or tissue-destructive process. In horses, hepatic amyloidosis occurs
chiefly as a result of chronic inflammation and has been well recognized in horses
used for the production of hyperimmune serum. Horses may develop icterus and other
signs of hepatic failure, but cattle die first of the primary disease or from uremia
resulting from concurrent renal amyloidosis. Dogs and cats typically develop signs
of renal dysfunction, although cats may be presented with spontaneous hepatic rupture.
Familial AA amyloidosis is recognized in Chinese Shar-Pei dogs. The condition resembles
familial Mediterranean fever in humans, and is characterized by febrile episodes,
swollen hock syndrome, and the development of renal and sometimes hepatic amyloidosis.
Familial AA amyloidosis is also recognized in Abyssinian cats and suspected in Siamese
and Oriental cats.
Affected livers in horses are pale, enlarged with rounded edges, friable, and prone
to fracture; in cattle, affected livers may be firm. Affected livers in all species
are predisposed to rupture and bleeding (eFig. 2-8). Amyloid is deposited first in
the parenchyma about the portal tracts and appears gray and waxy. Amyloid is deposited
in the perisinusoidal space between the sinusoidal lining and hepatocytes and is sometimes
found in the walls of the afferent vessels. The surrounded hepatocellular cords atrophy.
On H&E staining, amyloid appears as homogeneous eosinophilic amorphous extracellular
material (Fig. 2-27
). Staining with Congo red results in apple-green birefringence when viewed with polarized
light. Thioflavine T stain produces yellow-green fluorescent staining of amyloid when
viewed under ultraviolet light.
Figure 2-27
Amyloid in spaces of Disse compressing hepatocytes in a cat.
eFigure 2-8
Amyloidosis. A large, pale liver with capsular rupture and hemorrhage in a cat with
amyloidosis.
(Courtesy J.L. Caswell.)
Further reading
Armstrong PJ, Blanchard G. Hepatic lipidosis in cats. Vet Clin North Am Small Anim
Pract 2009;39:599-616.
Armstrong PJ, Williams DA. Pancreatitis in cats. Top Companion Anim Med 2012;27:140-147.
Badylak SF, Van Vleet JF. Tissue gamma-glutamyl transpeptidase activity and hepatic
ultrastructural alterations in dogs with experimentally induced glucocorticoid hepatopathy.
Am J Vet Res 1982;43:649-655.
Beatty JA, et al. Spontaneous hepatic rupture in six cats with systemic amyloidosis.
J Small Anim Pract 2002;43:355-363.
Bobe G, et al. Invited review: pathology, etiology, prevention, and treatment of fatty
liver in dairy cows. J Dairy Sci 2004;87:3105-3124.
Brix AE, et al. Glycogen storage disease type Ia in two littermate Maltese puppies.
Vet Pathol 1995;32:460-465.
Cebra CK, et al. Hepatic lipidosis in anorectic, lactating Holstein cattle: a retrospective
study of serum biochemical abnormalities. J Vet Intern Med 1997;11:231-237.
Center SA, et al. Ultrastructural hepatocellular features associated with severe hepatic
lipidosis in cats. Am J Vet Res 1993;54:724-731.
Center SA, et al. A retrospective study of 77 cats with severe hepatic lipidosis:
1975-1990. J Vet Intern Med 1993;7:349-359.
Dannatt L, Porter TA. An outbreak of ovine white liver disease in south west England.
Vet Rec 1996;139:371-373.
Downs-Kelly E, et al. Caprine mucopolysaccharidosis IIID: a preliminary trial of enzyme
replacement therapy. J Mol Neurosci 2000;15:251-262.
Gregory BL, et al. Glycogen storage disease type IIIa in curly-coated retrievers.
J Vet Intern Med 2007;21:40-46.
Haskins ME, et al. Hepatic storage of glycosaminoglycans in feline and canine models
of mucopolysaccharidoses I, VI, and VII. Vet Pathol 1992;29:112-119.
Helman RG, et al. The lesions of hepatic fatty cirrhosis in sheep. Vet Pathol 1995;32:635-640.
House JK, et al. Hemochromatosis in Salers cattle. J Vet Intern Med 1994;8:105-111.
Jakowski RM. Right hepatic lobe atrophy in horses: 17 cases (1983-1993). J Am Vet
Med Assoc 1994;204:1057-1061.
Johnstone AC, et al. The pathology of an inherited hyperlipoproteinaemia of cats.
J Comp Pathol 1990;102:125-137.
Kennedy S, et al. Histopathologic and ultrastructural alterations of white liver disease
in sheep experimentally depleted of cobalt. Vet Pathol 1997;34:575-584.
Kishnani PS, et al. Canine model and genomic structural organization of glycogen storage
disease type Ia (GSD Ia). Vet Pathol 2001;38:83-91.
Lee FY, et al. Activation of the farnesoid X receptor provides protection against
acetaminophen-induced hepatic toxicity. Mol Endocrinol 2010;24:1626-1636.
Loeven KO. Hepatic amyloidosis in two Chinese shar pei dogs. J Am Vet Med Assoc 1994;204:1212-1216.
Macleod NS, Allison CJ. Hepatic lipodystrophy of pedigree Galloway calves. Vet Rec
1999;144:143-145.
McCaskey PC, et al. Accumulation of 2,8 dihydroxyadenine in bovine liver, kidneys,
and lymph nodes. Vet Pathol 1991;28:99-109.
Mogg TD, Palmer JE. Hyperlipidemia, hyperlipemia, and hepatic lipidosis in American
miniature horses: 23 cases (1990-1994). J Am Vet Med Assoc 1995;207:604-607.
Munson L, et al. Diseases of captive cheetahs (Acinonyx jubatus jubatus) in South
Africa: a 20-year retrospective survey. J Zoo Wildl Med 1999;30:342-347.
Niewold TA, et al. Familial amyloidosis in cats: Siamese and Abyssinian AA proteins
differ in primary sequence and pattern of deposition. Amyloid 1999;6:205-209.
Ohlenbusch PD. A possible relationship between climate, vegetation, management and
occurrence of hard yellow liver disease. Vet Hum Toxicol 1990;32:583.
O'Toole D, et al. Hepatic failure and hemochromatosis of Salers and Salers-cross cattle.
Vet Pathol 2001;38:372-389.
Pearson EG, et al. Hepatic cirrhosis and hemochromatosis in three horses. J Am Vet
Med Assoc 1994;204:1053-1056.
Sanchez CR, et al. Use of desferoxamine and S-adenosylmethionine to treat hemochromatosis
in a red ruffed lemur (Varecia variegata ruber). Comp Med 2004;54:100-103.
van der Linde-Sipman JS, et al. Fatty liver syndrome in puppies. J Am Anim Hosp Assoc
1990;26:9-12.
van der Linde-Sipman JS, et al. Generalized AA-amyloidosis in Siamese and Oriental
cats. Vet Immunol Immunopathol 1997;56:1-10.
Whitney MS, et al. Ultracentrifugal and electrophoretic characteristics of the plasma
lipoproteins of miniature schnauzer dogs with idiopathic hyperlipoproteinemia. J Vet
Intern Med 1993;7:253-260.
Wilkerson MJ, et al. Clinical and morphologic features of mucopolysaccharidosis type
II in a dog: naturally occurring model of Hunter syndrome. Vet Pathol 1998;35:230-233.
Xenoulis PG, Steiner JM. Lipid metabolism and hyperlipidemia in dogs. Vet J 2010;183:12-21.
Yamazaki Y, et al. Role of nuclear receptor CAR in carbon tetrachloride-induced hepatotoxicity.
World J Gastroenterol 2005;11:5966-5972.
Types and Patterns of Cell Death in the Liver
Irreversible damage leads to cell death that is evident histologically as long as
the affected animal survives for a sufficient period of time, usually hours. Although
all cells in the liver are susceptible, most interest and evaluation has been directed
toward hepatocytes, biliary epithelium, and sinusoidal endothelium. The 2 predominant
types of cell death are necrosis, also termed oncotic necrosis, and apoptosis, although
there are other less common forms of cell death. These types of cell death can be
separated mechanistically and histologically to a certain extent.
Types of cell death
In routine diagnostic settings, the terms apoptosis and necrosis can be based on descriptive
criteria recognized in routine H&E-stained sections of liver. There have been attempts
to refine the terminology and criteria for cell death in the liver, but this is still
hindered by our incomplete understanding of different cell death pathways and cellular
responses in various diseases.
Apoptosis
Apoptosis
is a form of programmed cell death that permits removal of cell debris without much
leakage of cell contents or inflammation. Key features of apoptosis include retention
of plasma membrane integrity until the later stages of the process; proteolysis of
intracellular cytoskeletal proteins by aspartate-specific proteases, leading to collapse
of subcellular components; chromatin condensation and marginalization; nuclear fragmentation;
plasma membrane bleb formation; and eventual cell fragmentation into smaller apoptotic
bodies bound by intact plasma membrane (Fig. 2-28
). These bodies are rapidly phagocytosed and degraded by neighboring hepatocytes or
Kupffer cells. The rapid disappearance of these fragments means that very few apoptotic
bodies in a section can indicate a rapid rate of hepatocellular death and turnover.
Biochemically, apoptotic cells are characterized by phosphatidylserine on the outer
surface of the cell membrane, increased mitochondrial permeability with release of
internal proteins and activation of caspases.
Figure 2-28
Three apoptotic hepatocytes (arrows) in a region of injury in the liver of a dog.
Apoptosis is the main means of physiologic removal of damaged or aged cells and for
remodeling of tissue. Liver mass is maintained by a balance of mitosis and apoptosis.
Experimental support for the importance of this balance comes from studies of mutant
mice deficient in the principal mediator of apoptosis, the receptor Fas (Fas/CD95),
which develop prominent hepatocellular hyperplasia over time.
Apoptosis can be initiated by extrinsic or intrinsic events involving the death receptors
or mitochondria, respectively. Extrinsic mechanisms can be activated via cell surface
receptors termed death receptors when they bind their cognate ligands. Death receptors
include Fas, tumor necrosis factor-α (TNF-α) receptor and death receptor 4 (DR4) and
5 (DR5). The main signaling molecules in the liver are the Fas ligand (FasL), TNF-related
apoptosis-inducing ligand (TRAIL), and TNF-α. When the membrane-bound death receptors
bind their ligand, they trigger formation of a multiprotein complex, the death-inducing
signaling complex (DISC). Conformational changes in this complex trigger the activation
of caspase 8, the key mediator of apoptosis and subsequent activation of the effector
caspases: caspases 3, 6, and 7. More details on hepatocellular apoptosis are available
in recent reviews.
Intrinsic mechanisms involve mitochondrial injury with release of proapoptotic factors.
Activators of the latter pathway include oxidative stress, DNA damage, toxins, endoplasmic
reticulum (ER) stress, including the unfolded protein response, lipid peroxidation,
ultraviolet and γ-irradiation, and deprivation of growth factors. All of the pathways
initiated by these agents converge on the mitochondria, leading to mitochondrial outer
membrane permeabilization (MOMP). Mitochondrial membrane permeability is effected
either through direct membrane damage, or more commonly by opening of regulated transmembrane
pores by bax and other proapoptotic members of the Bcl-2 family. Cytochrome C and
other intermembrane mitochondrial factors released from the mitochondrion form an
apoptosomal complex in the cytoplasm that activates the distal caspase pathways, including
caspase 9, that execute many of the cell fragmentation events. Alternatively, MOMP
can be triggered by another mechanism involving multiprotein channels within the mitochondria,
leading to swelling and release of proapoptotic molecules.
Functional classification of cell death has been developed on the basis of chemical
rather than the histologic-morphologic criteria because different pathways can lead
to similar morphologies. For example, intrinsic pathways can be divided into caspase-dependent
and caspase-independent pathways.
Injury to the ER via oxidative stress, hypoxia, calcium depletion, inflammation, and
altered glycosylation, among other mechanisms, leads to the “unfolded protein response
(UPR)” that can also activate the distal caspase pathway by a separate mechanism.
Hepatocytes are also susceptible to an intrinsic mechanism of apoptosis that is active
after removal of hepatotrophic influences, including those that function via the constitutive
androstane receptor, and other nuclear receptors. In addition, lysosomes can undergo
selective membrane permeabilization, leading to a partial release of their contents
in response to death signaling mediated by oxidative stress, some lipid mediators,
and by Bcl-2 family members. Release of lysosomal proteases can cooperate with the
caspase cascade or act in a caspase-independent fashion.
Both extrinsic and intrinsic pathways lead, in turn, to disruption of the cytoskeleton
and nucleus primarily through the activation and direct action of caspases 3, 6, and
7. An important feature of apoptosis is the requirement for adenosine triphosphate
(ATP) to initiate the execution phase. If the degree of mitochondrial damage is sufficient
to exhaust ATP stores, membrane ion homeostasis deteriorates and leads to a mechanism
of cell death more consistent with necrosis. For example, although mild to moderate
oxidative stress may initiate intrinsic pathways of apoptotic cell death, processes
leading to marked oxidative stress typically cause cell death by necrosis, not only
resulting from the severity of mitochondrial damage, but also by direct inhibition
of the proapoptotic caspase cascade.
Loss of cell anchorage to the extracellular matrix can activate a form of programmed
cell death related to apoptosis that is termed
anoikis. Detached cells can die intact, as in exfoliation, or undergo typical fragmentation
into apoptotic bodies. Hepatocyte survival in vitro depends on their integrins that
adhere to the extracellular matrix, but it is still unknown how loss of these contributes
to cell death of hepatocytes in the intact liver.
Phagocytosis of apoptotic fragments follows flipping of phosphatidylserine from the
inner leaflet of the plasma membrane to the outer leaf, where it can then be recognized
by the phosphatidylserine scavenger receptor. Various assays can be used to detect
portions of these pathways in hepatocytes; caspase-cleaved cytokeratin 18 can be recognized
by antibodies to an internal domain, whereas chromatin fragmented by a calcium-activated
endonuclease generates double-strand breaks that can be tagged by the terminal deoxynucleotide
transferase-mediated dUTP nick-end labeling (TUNEL) technique. However, there are
still few diagnostic situations in which these indicators have been shown to be useful.
Increased numbers of apoptotic hepatocytes can be more readily assessed with a suitable
cytologic marker. In routine sections, a previous increase in apoptosis can be suggested
by increased amounts of lysosomal debris and pigment in Kupffer cells, but this is
not specific for apoptosis.
Rapid phagocytic removal of apoptotic hepatocyte fragments can minimize secondary
inflammation in cases of minor injury. However, phagocytosis of apoptotic bodies by
Kupffer cells and hepatic stellate cells is not innocuous and can engender inflammation
and fibrosis. Kupffer cells and resident macrophages express several ligands (TNF-α,
TRAIL, and Fas ligand) that lead to increased hepatocyte apoptosis. Similarly, hepatic
stellate cells are stimulated to release profibrotic cytokines and type 1 collagen
following phagocytosis of apoptotic bodies. In addition, apoptotic cells release nucleotides
ATP and uridine triphosphate (UTP), which can bind to purinergic receptors on macrophages
and hepatic stellate cells and which may provide additional stimulus for fibrosis
and inflammation. Excessive apoptosis is currently viewed as a driver of hepatic inflammation
and fibrosis.
Necrosis
Necrosis
is morphologically recognizable by initial cell swelling and subsequent loss of plasma
membrane integrity, leading to large organelle-free blebs and cell lysis. Some authors
prefer the term “oncosis” to emphasize the cell swelling response in necrosis to contrast
with cell shrinkage in apoptosis (Fig. 2-29
). Necrosis occurs when cells are depleted of ATP and lack sufficient energy to maintain
membrane associated ionic pumps, leading to swelling and gross calcium influxes that
result in disruption of the mitochondrial and plasma membranes, including release
of lysosomal enzymes. Oncosis is generally regarded as a severe injury to cell membrane
integrity or other vital functions, leading to enzyme leakage, inflammation, and tissue
repair. Release of cellular contents typically elicits a secondary inflammatory response
and provides the serum enzymes that are useful clinically to detect liver necrosis.
However, these sequelae can also occur after hepatocyte apoptosis, either because
the capacity for phagocytic removal of dead cells is exceeded, or because the insult
worsens such that necrosis may supervene.
Figure 2-29
Necrosis of hepatocytes is initiated by cell swelling evident in this toluidine blue–stained
section of liver.
(Courtesy V. Meador)
Necrosis is often a consequence of profound loss of mitochondrial function involving
the opening of the membrane permeability transition (MPT) pore, a megachannel composed
of inner- and outer-membrane proteins. This results in mitochondrial depolarization
and ATP depletion because of inhibited oxidative phosphorylation. Poly-ADP-ribose
polymerase (PARP) is a DNA repair enzyme that can deplete injured cells of available
ATP when it is activated to repair the multitude of DNA strand breaks induced by cell
damage. Conversely, PARP is swiftly cleaved in apoptosis to maintain ATP levels. Much
of the ATP in hepatocytes is used for membrane homeostasis of Na+, K+, and Ca2+ ions.
ATP depletion or physical damage to membranes of the cell surface, mitochondria, and
ER leads to loss of ion homeostasis, altered cellular volume regulation, and increased
intracellular calcium ion concentrations. Activation of calcium-dependent endonucleases,
proteases (e.g., calpains), and phospholipases is responsible for the terminal events
in necrosis. The source of calcium may influence the type of cell death, as calcium
influx from the plasma membrane is associated with necrosis, but calcium released
from the ER is more likely to trigger apoptosis. Newer evidence suggests that necrosis
is not entirely a passive process. For example, necrosis can be initiated via cell
surface receptor–driven processes when TNF is present in high concentrations.
The term
coagulative necrosis
may be applied to groups or zones of intact, but dead hepatocytes that have shrunken
slightly, stain intensely with eosin, and may have visible but distorted nuclei (Fig.
2-30
). These cells may also be dehydrated but, unlike apoptosis, the removal of water
is not an active process, and the affected cells do not undergo spontaneous fragmentation.
It seems that coagulative necrosis, which is often seen in acute hepatotoxicity, is
the result of sudden and catastrophic denaturation of cytosolic protein, which imparts
a rather dense, rigid texture to the dead cells, somewhat preserving their shape.
The term
lytic necrosis
has been used for areas of necrosis in which the hepatocytes are disintegrating, usually
in the presence of infiltrating phagocytes, especially neutrophils. This is consistent
with the later stages of postnecrotic inflammation that follow necrosis rather than
apoptosis.
Figure 2-30
Coagulative necrosis of infarcted hepatocytes in a cat resulting from compression
by an adjacent mass.
One might expect that the different types of cell death might be informative in relation
to causes and pathogenesis. However, apoptosis, necrosis, and mixed responses are
common in liver injury in vivo. Many insults can initiate either necrosis or apoptosis
of hepatocytes; these include hypoxia, reactive oxygen metabolites, hepatotoxic chemicals,
viral infections, bacterial toxins, and inflammation. Some of the molecular events
are common to both. Mitochondrial damage and several other activation responses are
common to apoptosis and necrosis, and programmed cell death responses can be activated
before they are overwhelmed by more intense injury responsible for necrosis. Susceptibility
therefore depends on many factors, including the level and duration of insults, replication
status, and integrity of the various homeostatic and cytoprotective functions. Indeed,
review of caspase-cleaved substrates reveals that different toxic drugs do not cause
a uniform pattern of caspase activation and that it is likely that there are distinct
pathophysiologic pathways of apoptosis. Clearly, apoptosis is not a consistent and
stereotypic response.
There are important misconceptions regarding differences between apoptosis and necrosis
and their effects on the liver. It is often assumed that apoptosis, unlike necrosis,
leads to a “clean” death that does not provoke inflammation or elevation of transaminases
and other markers of inflammation. However, there is evidence that phagocytosis of
apoptotic bodies, termed efferocytosis, particularly by Kupffer cells can, in fact,
activate the Kupffer cells and stimulate additional cell death. Various inflammatory
markers are elevated in both acute and chronic apoptosis. The view that apoptosis,
because it involves release of intact membrane bodies, does not lead to transaminase
elevation has been shown to be incorrect in several studies. Injection of Fas ligands
has been shown to produce significant increases in serum transaminases within hours
and that this response is blocked in Bcl-2 transgenic mice. In addition, deletion
of anti-apoptotic proteins results in elevated transaminase levels.
Other forms of cell death
There are a number of other forms of cell death. These include autophagy, pyroptosis,
necroptosis, entosis, netosis, and parthanatos, which can be identified by biochemical
means and specific forms of inhibition. A complete description is beyond the scope
of this text.
Tissue patterns of cell death
Focal necrosis
Focal necrosis is very common in autopsy material. The lesions are microscopic or
barely visible to the naked eye and are usually numerous. Their designation as focal
depends on their size and on a random distribution relative to the lobules. There
may be a tendency for focal necrosis to occur nearer to the portal vessels than to
the periphery of the circulatory fields, and to be concentrated in some lobular agglomerates
rather than others.
Focal necrosis occurs in many infections, parasitic migrations, and instances of biliary
obstruction, and in these, the designation focal hepatitis will often be more appropriate,
because most are attended by some degree of focal inflammation. The infectious causes
may be viral (Fig. 2-31
) or bacterial. Many septicemic bacterial infections consistently produce focal hepatic
lesions; examples are salmonellosis, tularemia, pseudotuberculosis, listeriosis in
the fetus and newborn, and Mannheimia haemolytica septicemia in lambs. The focal necrosis
may be the outcome of a Kupffer cell reaction, as in salmonellosis, or of bacterial
embolism. The cause can usually be determined by histologic examination.
Figure 2-31
Focal necrosis caused by canid herpesvirus 1 in a dog.
In cattle, focal necrosis in a few or many visible foci is common at autopsy and is
common enough to be important at slaughter; it is responsible for the descriptive
appellation “sawdust liver” (Fig. 2-32
). The pathogenesis is not known and probably varies, but it may be caused by organisms
from the gut that reach the liver in the portal blood. The lesion is not specific
and consists of focal parenchymal necrosis with disruption of reticulin fibers and
infiltration of neutrophils and lymphocytes; frank suppuration does not occur. This
lesion is said to be more frequent in livers from feedlot-fattened cattle.
Figure 2-32
Focal hepatitis—the so-called “sawdust liver” of cattle.
Focal necrosis in biliary obstruction follows rupture of distended canaliculi or smaller
cholangioles, with the formation of small bile lakes. Necrosis of larger bile ducts
caused by neutrophilic cholangitis or destructive cholangitis can also release bile.
The yellow pigment is readily visible microscopically and provokes small granulomas
with giant cells.
Focal necrosis is of very little functional significance for the liver, even when
numerous. The lesions heal with some scarring, but this too probably disappears in
time. They are of diagnostic importance in some diseases such as salmonellosis, and
as indicators of possible bacteremia for meat inspectors.
Lobular necrosis
Although various terms are used, the simplest way to visualize these patterns of necrosis
is by using the hepatic lobule concept.
The time-honored designation “centrilobular necrosis” is a common lesion in response
to intoxication or hypoxia. Centrilobular (zone 3) necrosis
is the most common form of zonal necrosis in domestic animals. The hepatocytes in
the centrilobular zone are particularly vulnerable to necrosis, in part because they
are farthest from incoming arterial and portal venous blood bearing oxygen and essential
nutrients. They also contain the greatest concentration of cytochromes P450 that activate
various exogenous compounds into reactive metabolites capable of injuring or killing
hepatocytes (see later section on Toxic hepatic disease).
Severe viral infections, such as canine adenovirus 1 and Rift Valley fever virus,
can produce centrilobular necrosis, and the reasons for the increased susceptibility
of the hepatocytes of this zone in these diseases are not established. Plausible explanations
include zonal expression of entry receptors used by viruses to infect hepatocytes
or ischemia-related hepatocellular swelling and sinusoidal damage because of virus-induced
endothelial injury that reduce effective perfusion of the centrilobular hepatocytes.
Centrilobular degeneration and necrosis are seen commonly in animals that have died
rather slowly. It is assumed that, in the agonal period, the hepatocytes in this zone
are disproportionately affected by tissue hypoxia as a result of the failing circulation.
This necrosis is more extensive if the animal is anemic. Centrilobular necrosis is
also seen in passive venous congestion of the liver, most often in cases of acute
and dramatic decompensation of cardiac function, as atrophy of hepatocytes is a more
common outcome.
In the liver with centrilobular necrosis, there is usually a prominent zonal pattern,
which takes the form of a fine, regular, pallid network of surviving, often fatty,
hepatocytes in the periportal zone, which stands up above the red, collapsed areas
adjacent to the hepatic venules (Fig. 2-33
). The zonal necrotic insult frequently affects the sinusoidal endothelium, allowing
erythrocytes to enter the perisinusoidal space and contribute to the redness of the
necrotic zones. Histologically, necrosis is confined to the centrilobular region (Fig.
2-34
), although bridging can occur in more severe cases (Fig. 2-35
). Necrotic cells that are removed can be replaced by stagnant blood, at least in
the acute phase. However, this red-on-yellow zonal pattern is difficult to interpret
grossly because hepatic steatosis may also have a zonal distribution without having
significant necrosis.
Figure 2-33
Enhanced zonal pattern of hepatocellular necrosis in a horse.
(Courtesy R. Panciera.)
Figure 2-34
Acute centrilobular necrosis in an ox.
Figure 2-35
Bridging centrilobular hemorrhagic necrosis caused by Xamia sp. poisoning in a sheep.
However, vascular influences on the susceptibility to necrosis mean that segments
of the lobule can be differentially affected. Frequently, the areas of necrosis are
joined to one another, thus cutting the conventional lobules into segments, and at
the same time, outlining the periphery of the circulatory fields of the hepatic lobules.
Some of these areas of necrosis extend up to larger portal triads, because the periphery
of some lobules may lie against the larger portal tracts. Often, the hepatocytes between
the necrotic and more normal zones show hydropic degeneration or fatty change.
If the insult is of short duration, quite extensive centrilobular necrosis may be
followed by phagocytic infiltration and hepatocellular proliferation and complete
restoration of normal structure and function within a few days. Severe centrilobular
necrosis may be followed shortly by proliferation of bipotential progenitor cells
found near cholangioles, termed the ductular reaction, and mature bile duct epithelia
that also respond to the regenerative stimulus. With restitution of the normal complement
of hepatocytes, the proliferative response in the progenitor cells and biliary tract
subsides unless the original insult is continuous or repeated.
Some intoxications can produce selective
midzonal (zone 2) necrosis, affecting only a narrow, sharply defined band of hepatocytes
(Fig. 2-36
). It may be more diffuse within the lobule, so that periportal or centrilobular degeneration
may be superimposed on the more severe midzonal lesion.
Figure 2-36
Acute midzonal necrosis in a foal.
Periportal (zone 1) necrosis
is also an uncommon lesion, perhaps more often seen than midzonal necrosis, and can
be caused by direct-acting hepatotoxins that do not require metabolism by the cytochromes
P450 to produce toxic moieties. More than one pattern of zonal necrosis can be found
in the same liver. The various forms of zonal necrosis cannot reliably be distinguished
from one another grossly, but one may expect to see in periportal necrosis a reversal
of the pattern seen in centrilobular necrosis; that is, in periportal necrosis, the
surviving hepatocytes about the hepatic venules may appear as pale, raised islands
within a regular network of red, collapsed periportal tissue. Careful scrutiny may
reveal the smallest hepatic venules at the center of the pale islands.
Paracentral necrosis,
a form of coagulative necrosis, occurs when an isolated portion of the centrilobular
region of the lobule (or complete hepatic acinus of Rappaport—perhaps visualized more
easily as an entire acinus) dies and is viewed in transverse section (Fig. 2-37
). It is possibly an ischemic lesion or infarct produced by an occlusion of a terminal
portal venule, such as may occur in disseminated intravascular coagulation. Its appearance
in certain of the acute hepatotoxicities probably represents the death of a single
complete acinus as a result of local vascular insufficiency, although, theoretically,
high local microsomal enzyme activity or local deficiency of hepatocellular protective
factors may play a part. Occlusion and rupture of a bile ductule or cholangiole are
other potential causes of paracentral necrosis.
Figure 2-37
Paracentral necrosis.
Massive necrosis
Massive necrosis refers to necrosis of an entire hepatic lobule, not necessarily necrosis
of the liver as a whole. By accepted definition, every cell in the affected lobule
is dead, including the hepatocytes of the limiting plate (Fig. 2-38
). Without surviving parenchyma to support regeneration, affected lobules collapse,
so that portal areas and hepatic venules are approximated and the intervening stroma
is condensed. Such a liver must regenerate from surviving hepatocytes in other, less
severely affected lobules or by proliferation of progenitor cells that mature into
hepatocytes. The distribution of massive hepatic necrosis often relates to the distribution
of larger vessels. Collapse, condensation, and subsequent scarring are characteristic,
the end result being known as postnecrotic scarring. The liver is not uniformly involved
typically. Large areas of parenchyma remain intact, and these enlarge amid scarring
areas during compensatory regeneration.
Figure 2-38
Massive necrosis with destruction of the periportal limiting plate and ductular reaction
caused by Amanita poisoning in a cat.
A liver that is the seat of massive necrosis may be of normal size or smaller. Fine
red threads of fibrin may be present on the surface, especially in the grooves between
the lobes. There is a surface mosaic appearance of red, gray, or yellow areas intermingled
with areas of dark red. The gray or yellow areas of parenchyma, representing surviving
tissue, form irregular, coalescing patches that may be <1.0 cm in diameter. The intermingled
red areas represent areas of necrosis, hemorrhage, and collapse, and these are depressed
a few millimeters below the surface. In the healing stage, the depressed areas of
hemorrhage and necrosis are condensed, shrunken, and scarified so that the surface
of the liver is traversed by fine or heavy scars that separate large nodules of regenerative
hyperplasia. Further acute episodes may be superimposed so that the presented lesion
may be a mixture of acute massive necrosis and postnecrotic scarring.
Hepatosis dietetica
of swine is a now uncommon syndrome of massive hepatic necrosis in association with
its immediate or late effects, namely “yellow-fat disease,” degeneration of skeletal
and cardiac muscle, serous effusions, ulceration of the squamous mucosa of the stomach,
and fibrinoid necrosis of arterioles. These lesions may occur alone or in any combination,
although all seldom occur in one animal. They are known to be of nutritional origin,
and the fact that the various lesions can occur separately indicates the complexity
of the pathogenesis. Experimental observations have revealed the need for concurrent
deficiencies of sulfur-containing amino acids, tocopherols, and trace amounts of selenium
if hepatic necrosis is to develop. Selenium protects efficiently against the hepatic
necrosis and massive effusions, and tocopherols are probably protective against other
lesions that occur as part of the syndrome. The pathogenesis is incompletely understood,
but is in part related to the generation of free radicals, exacerbated by deficiency
of free-radical scavengers such as vitamin E, and selenium, protective against reactive
oxygen radicals through its role in glutathione peroxidase and some other selenoproteins.
Hepatosis dietetica occurs in rapidly growing pigs fed diets largely of grain and
containing protein supplements lacking in either quality or quantity. There is some
evidence that in pigs that are nutritionally predisposed, a cold and damp environment
or some other stress may precipitate the disease. Death usually occurs without signs
of illness or after a short period of dullness. Melena, dyspnea, weakness, and trembling
may be observed in some cases. Jaundice is indicative of a relapsing course.
Affected pigs are usually in good condition. The carcass may be anemic if ulceration
of the gastric mucosa has occurred, and, in these cases, free and digested blood may
be found in the stomach and intestine. Jaundice is not common, but yellow staining
of adipose tissues (yellow-fat disease) is. In relapsing cases, hemorrhagic diathesis
may occur, manifested mainly by hemorrhage into and about joints. Protein-rich fluid
collects in the serous cavities in small volume. Fine strands of fibrin are present
in the peritoneal cavity.
Pulmonary edema accompanies myocardial lesions that consist of intramural and subendocardial
hemorrhages with focal areas of hyaline degeneration. The changes in the liver dominate
the autopsy findings. The massive hepatic necrosis is of the typical appearance described
earlier, and in a number of cases, both acute and chronic lesions are found (Fig.
2-39
). The sites of severest injury are the dorsal parts on the diaphragmatic surface.
The right lobe may escape and later undergo marked hypertrophy. The gallbladder is
often edematous.
Figure 2-39
Hepatosis dietetica in a pig.
(Courtesy University of Guelph.)
The histologic changes that occur in this syndrome are described elsewhere with the
particular organs involved. Briefly, centrilobular necrosis of the liver is typical
(eFig. 2-9). Additionally, fibrinoid degeneration of small arteries occurs in some
cases. The arterial degeneration may occur in any organ or in most organs but is relatively
common in only the small vessels of the mesentery, gut, and heart (see Mulberry heart
disease, in Vol. 3, Cardiovascular system).
eFigure 2-9
Centrilobular necrosis in hepatosis dietetica.
Piecemeal necrosis
Models of immune-mediated hepatocyte necrosis have emerged from studies of human viral
hepatitis and some forms of drug-induced chronic hepatitis. This pattern of necrosis
amid sites of more active inflammation at the limiting plate of hepatocytes immediately
adjacent to the edge of the portal tract is referred to as piecemeal necrosis. The
mechanisms may involve either direct damage to hepatocytes by the uptake of antigen-antibody
complexes, or cooperation between macrophages and T lymphocytes. These may cause cell-mediated
destruction of hepatocytes that have taken up these complexes or, perhaps, native
antigen or virus. Whatever the agency, the mode of cell removal in this sort of injury
often takes the form of single-cell necrosis or apoptosis, which may be directly triggered
by immunologically competent cells. This type of liver injury, in which inflammation
characteristically disrupts the limiting plate, giving an irregular appearance to
the periportal zone, is discussed further in the section on Chronic hepatitis.
Necrosis of sinusoidal lining cells
Injury to the endothelium of hepatic sinusoids can develop in a variety of forms.
The sinusoidal endothelial cells may lose their typical fenestrae or pores, limiting
contact between plasma and hepatocellular microvilli, and if there is an increased
deposition of extracellular matrix (ECM) beneath the altered endothelium, the alteration
is termed “capillarization of sinusoids,” leading to reduced hepatic function. Sinusoidal
endothelial cells may relax their connections to the fine meshwork of the hepatic
“reticulin” or detach completely forming microemboli in the sinusoid downstream. In
addition, the residual, exposed space of Disse has an increased risk of sinusoidal
thrombosis. Acute endothelial injury can occur as a result of ischemia-reperfusion
injury, acetaminophen or pyrrolizidine toxicity, and effects of endotoxin, all of
which can lead to localized ischemia and necrosis. Other endothelial toxins include
ngaione poisoning and microcystin-LR, a highly toxic cyclic heptapeptide produced
by Microcystis aeruginosa, an aquatic cyanobacterium (blue-green alga). Microcystin-LR
is selectively injurious to hepatic sinusoidal endothelial cells by inhibiting protein
phosphatases. When the cytoskeleton becomes hyperphosphorylated, endothelial cells
undergo apoptosis, leading to hemorrhagic necrosis.
Some forms of peliosis hepatis may develop following endothelial necrosis. More chronic
injury can produce fibrosis, leading to diminished or disrupted sinusoidal blood flow.
Alteration of the sinusoidal blood flow can be a primary event leading to hepatocyte
hypoxia with liver dysfunction and disruption of the portal circulation. Secondary
endothelial cell injury can result when hepatocytes are being destroyed by the elaboration
of toxic molecules within their cytoplasm, it is to be expected that the sinusoidal
lining cells may also suffer should the products of these biotransformations spill
into the space of Disse. Erythrocytes increase in the perisinusoidal space and, when
endothelial damage is severe, the regions of necrosis are hemorrhagic. Necrosis of
sinusoidal endothelial cells is seen very early in the course of many hepatotoxicities.
Sinusoidal phagocytes are sometimes vulnerable to necrosis by virtue of their role
in clearing the portal blood of particulate or colloidal material; should these particles
be toxic or infectious, Kupffer cell necrosis may occur alone, but more usually, there
is damage to surrounding hepatocytes as well.
Necrosis of bile duct epithelium
It is unusual for the bile duct epithelium to be singled out by specific lethal insults,
but this is seen in intoxication by sporidesmin (see section on Toxic hepatic disease).
Usually, there is accompanying portal inflammation. Some experimental toxicants, such
as α-naphthylisothiocyanate, are lethal to bile duct epithelia and elicit a local
inflammatory cholangitis. Idiosyncratic drug-induced destructive cholangitis can lead
to acute cholangiolar injury as well as chronic cholestasis with damage and loss of
bile ducts (destructive cholangitis). Treatment with sulfonamides, other drugs, and
viral infection have been linked to this response (Fig. 2-40
).
Figure 2-40
Acute necrosis of biliary epithelium in a dog following exposure to trimethoprim-sulfa.
Further reading
Copple BL, et al. Endothelial cell injury and fibrin deposition in rat liver after
monocrotaline exposure. Toxicol Sci 2002;65:309-318.
Guicciardi ME, et al. Apoptosis and necrosis in the liver. Compr Physiol 2013;3:977-1010.
Luedde T, et al. Cell death and cell death responses in liver disease: mechanisms
and clinical relevance. Gastroenterol 2014;147:765-783.
Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011;147:728-741.
Responses of the Liver to Injury
The liver is a remarkably versatile organ. Given its central position in the body,
supplied by blood draining the gastrointestinal tract, and the fact that it is the
key organ involved in detoxification of exogenous (xenobiotics) and endogenous compounds
(endobiotics), the liver must respond to injury, including metabolic, infectious,
and hemodynamic insults. This section describes the 3 main categories of liver response
to injury: regeneration, fibrosis, and ductular reaction (biliary hyperplasia).
Hepatic regeneration
Mild injury involving the hepatocytes, biliary epithelium, endothelial cells, and
mesenchymal elements typically is completely resolved quickly through regeneration.
This is particularly true in younger animals, as the vigor of regeneration diminishes
with age. Even in cases of massive hepatocellular necrosis, complete recovery is possible
if the reticulin framework of the liver persists and surviving hepatocytes have sufficient
time to regenerate and replace the lost hepatic mass.
In adulthood, the liver is a stable organ with limited replication of mature hepatocytes,
biliary epithelium, and other cells in the liver. However, loss of hepatic mass through
injury, such as infections, toxic insult, or surgical removal, will transform the
mature hepatocytes, biliary epithelium, and/or bipotential progenitor cells, as well
as hepatic stellate cells and endothelial cells, into actively proliferating cells
until normal functional mass has been replaced. As much as 70% of the normal liver
can be removed surgically without clinical insufficiency, and in the course of a few
weeks, it is back to its normal mass. Depending on the nature of the injury, regeneration
occurs via one of 2 pathways. In cases of moderate injury, cell loss is replaced by
proliferation of mature hepatocytes or biliary epithelium, providing swift replacement
of parenchymal loss. In cases of more severe injury, or conditions that inhibit replication
of mature cells, there is proliferation of bipotential progenitor cells, called a
ductular reaction. These cells reside in the periportal region of the lobule at the
level of the canals of Hering, the site where bile canaliculi and biliary epithelial
cells first join and form the beginning of the epithelium-lined biliary tree. They
are recognized histologically as small, basophilic cells that form single-cell arrays
or ductules with narrow lumens. Depending on the differentiation pathway that is followed,
they can mature into hepatocytes or bile duct epithelium.
Regeneration of mature hepatocytes following surgical removal is well known. An example
from Greek mythology is Prometheus, punished for giving fire to the mortals, daily
had his liver partially eaten by an eagle (a symbol of Zeus), only to regrow and be
eaten again. Although the mythical daily regrowth of hepatic mass is more prodigious
than the real-world response, hepatic regeneration is nonetheless remarkable. In rodents,
partial hepatectomies can be repeated more than 10 times, followed by restoration
of normal hepatic mass. Individual hepatocytes are able to replicate up to 70 times.
Simple replacement of missing hepatocytes alone does not truly constitute regeneration
because normal liver microscopic architecture is essential to maintain normal hepatic
function. Although new lobes are not regenerated following hepatectomy, the remaining
lobes enlarge, and the proliferated hepatocytes and other cellular elements progress
through a series of steps that involve interaction of hepatocytes and angiogenic processes
to restore the normal relationships between the hepatocytes and the sinusoids.
Regeneration of mature hepatocytes is stimulated by several factors. These include
polypeptide growth factors, such as hepatocyte growth factor, epidermal growth factor,
transforming growth factor-α (TGF-α), and insulin-like growth factors. Hepatocyte
growth factor is the most potent mitogen and is released by hepatic stellate cells
to stimulate hepatocytes in a paracrine fashion. This growth factor can also be embedded
in the ECM to be released by proteases. Hepatic regeneration is also enhanced by nutrition,
with fasting slowing and dietary protein promoting regeneration. In addition, catecholamines
and a broad variety of hormones support regeneration, with insulin, glucagon being
the most significant. Cytokines, such as IL-6, produced by TNF-α–stimulated Kupffer
cells, are other important effectors. When IL-6 binds its receptor, it stimulates
mitogenic intracellular signaling and potentiates signaling by other growth factors.
These effectors are examples of the important role of the innate immune system in
hepatic regeneration. The autonomic nervous system also plays a role in hepatic regeneration,
as denervation of the liver can significantly limit hepatic regeneration.
Following hepatocyte proliferation, normal liver architecture must be preserved to
maintain hepatic function. Several cell types are involved in this process. Following
partial hepatectomy, the initial proliferation creates aggregates of hepatocytes 10-14
cells thick. These aggregates are penetrated by endothelial buds that eventually establish
sinusoidal channels. Ingrowth of the endothelial cells is driven by regulators of
angiogenesis, such as vascular endothelial growth factor (VEGF) and platelet-derived
growth factor (PDGF). Hepatic stellate cells replicate and synthesize ECM within ~4
days of partial hepatectomy and help establish normal hepatocyte-matrix interactions.
Once the needed hepatic mass has been replaced, the regenerative process must be regulated
to terminate cell proliferation. TGF-β and related family members, including activin,
are the best known mediators responsible for curtailing hepatocyte proliferation.
These mediators are produced primarily by the hepatic stellate cells. They can increase
ECM production as well as suppress hepatocellular replication, are downregulated during
proliferation, and increase once appropriate hepatic mass has been achieved.
In severe, often toxic, liver injury, mature hepatocytes cannot replace damaged hepatic
mass, and bipotential progenitor cells are engaged to restore the hepatic mass. At
the interface of the portal tract connective tissue and the hepatic parenchyma, and
in the vicinity of the canals of Hering, there is a compartment that serves as the
site of quiescent progenitor cells. Proliferation of these cells gives rise to the
“ductular reaction,” characterized by ductular structures lined by cuboidal, basophilic
cells (Fig. 2-41A
). The relationship between the “oval cells” described in rodents and the ductular
reaction still requires some clarification, but it is likely that the same cell type
is involved. In addition, there may be progenitor cells among the mature biliary epithelial
cells, and others may be marrow derived and blood-borne to the liver. Immunohistochemical
staining of these cells in humans and rodents reveals that they contain cytokeratin
markers of biliary epithelium and, simultaneously, hepatocyte markers such as albumin
(Fig. 2-41B). Similar cells are present in animals as well. There are several circumstances
that can trigger the ductular reaction other than massive necrosis, although this
is the best recognized form. Both cholestasis and hypoxia can drive the ductular reaction
causing proliferation in the periportal region for the former, and in the centrilobular
region in the latter instance. The ability of these cells in the ductular reaction
to differentiate into mature biliary or hepatocellular phenotypes is well established.
It should be appreciated that in most clinical circumstances, hepatic regeneration
is a less orderly process than that which occurs in healthy experimental animals.
Regeneration requires normal arterial and venous inflow as well as venous and biliary
drainage. Without a supportive environment, hepatic regeneration of hepatocytes, biliary
epithelium, and nonparenchymal cells may be quite variable within different regions
of the liver. Following massive necrosis, all remaining hepatocytes are likely to
initiate a replicative response, whereas random focal necrosis will engender a local
proliferation of hepatocytes. Scattered individual cell necrosis is repaired almost
imperceptibly. In the more common pattern of centrilobular necrosis, seen in many
toxic insults, complete repair is usually evident with a 24-48 hour period. Chronic
or repetitive injury often results in nodule formation as discussed in the section
on Fibrosis.
Figure 2-41
A. Ductular reaction in an injured liver. B. Cytokeratin 7 stain.
Ductular reaction (bile duct hyperplasia)
Ductular reaction (biliary hyperplasia) is a characteristic reaction of the liver
to various types of insult involving proliferation of bile duct epithelium, hepatic
progenitor cells, and possibly metaplasia of mature hepatocytes. The histologic hallmark
of the ductular reaction is the formation of new, irregular, and tortuous ductules
or chains of cells formed from flattened or cuboidal basophilic epithelium.
There are 3 main types of ductular reactions. (1) A common association with acute
bile duct obstruction is proliferation of preformed bile duct epithelium, creating
a rapid increase in bile duct surface area. This can protect hepatocytes from bile
acid buildup through a recirculation process termed cholehepatic recirculation. (2)
In cases of serious injury to hepatocytes, particularly when mature hepatocytes are
not able to replicate, there is a prominent ductular reaction originating from hepatic
bipotential progenitor cells (or oval cells). The bipotential nature of these cells
is supported by the detection of biliary epithelial cytokeratins 7 and 19 in cells
with an otherwise hepatocellular phenotype. A dramatic presentation of ductular proliferation
is often evident in animals that succumb several days after a fatal hepatic intoxication
or in horses with Theiler's disease. (3) Ductular reaction may also result from metaplasia
of mature hepatocytes in regions of hypoxia.
Ductular reaction may also develop secondary to local portal inflammation and fibrosis.
Ductular formation can also occur in areas of hypoxia. Such ducts formed by metaplasia
of mature hepatocytes can be found in centrilobular areas in animals with chronic
heart failure and passive congestion, for example (Fig. 2-42
).
Figure 2-42
Ductule formation in the centrilobular region of a dog with chronic right-sided heart
failure.
However, reabsorption of bile constituents by biliary epithelium leads to a neutrophilic
inflammatory reaction and local edema formation, which can be confused with a bacterial
infection. With persistence, the ductular reaction can contribute to portal and periportal
fibrosis as the ductular cells secrete profibrogenic growth factors, cytokines, and
chemokines.
Illustrative examples from natural disease are provided by the toxicoses of phomopsin,
pyrrolizidine alkaloids, and aflatoxin, and by equine serum hepatitis and ovine white-liver
disease.
Fibrosis
Hepatic fibrosis is a potentially reversible form of wound healing in which there
is an accumulation of various ECM components. In cases of acute or self-limited injury,
fibrosis may resolve completely and normal liver architecture can be restored, but
when damage is too extensive to be repaired, a scar may be permanent, and if the injury
is persistent, there is progressive deposition of ECM, leading to the “final common
pathway” and ending in fibrosis and cirrhosis. In normal liver, the ECM exists in
a state of dynamic balance, with synthesis and degradation occurring in balance to
maintain normal levels of all constituents. It is found in the portal tracts, along
the sinusoids in the space of Disse, around the central vein and in Glisson's capsule.
Normally, the hepatic ECM constitutes only 3% of the relative areas on a tissue section
and only 0.5% of the total liver wet weight. The main constituents of the hepatic
ECM are collagen, proteoglycans, laminin, fibronectin, and matricellular proteins.
In the space of Disse, the matrix is composed primarily of collagens IV and VI. Following
injury, however, fibrillar collagens I and III as well as fibronectin are deposited.
These deposits disrupt the normal structure and function of the sinusoids, leading
to capillarization of sinusoids, discussed later. The ECM is not merely scaffolding
for the liver. It also functions as a repository for various growth factors and metalloproteinases
in latent form, and it can bind a variety of survival factors, such as hepatocyte
growth factor and TNF-α, that may protect the growth factors and inhibit hepatocyte
apoptosis. In addition, interactions between cells and the ECM can affect the phenotype
of the hepatocytes and stellate cells and the composition of the ECM. Thus the process
of hepatic fibrosis is distinct from the condensation of the hepatic reticular framework
that can be seen following hepatocyte loss.
The process of ECM production in the injured liver is initiated by a process that
activates hepatic mesenchymal cells into myofibroblasts. These mesenchymal cells constitute
a prototypical mesenchymal cell type that mediates injury and repair in a number of
tissues, including the kidney, skin, lung, and bone marrow. All of these cell types
have the ability to produce ECM, and have contractile ability. The best recognized
group in this family is the hepatic stellate cells, but there are other important
groups in this family, including portal fibroblasts, bone marrow–derived cells, and,
via epithelial-mesenchymal transition, hepatocytes that can acquire a mesenchymal
phenotype. Portal fibroblasts, particularly those surrounding the bile ducts, are
an important source of ECM formation in the portal tracts. This is particularly evident
in cases of biliary tree injury. Portal fibroblasts/myofibroblasts are distinct from
stellate cells based on distinct protein markers.
Hepatic stellate cells are found in the space of Disse, often between hepatocytes.
In normal circumstances, these cells store lipid, primarily vitamin A (retinyl esters),
in characteristic large round vacuoles. In injured liver, hepatic stellate cells become
activated. A principal morphologic change involves the loss of the lipid vacuole and
transformation into a spindle-shaped myofibroblast. In some species, stellate cells
contain immunohistochemically detectable α–smooth muscle actin before activation,
and in other species, activation is required for expression of this protein. When
activated, they replicate robustly at sites of injury and are the main source of collagen
and other ECM components, such as proteoglycans, fibronectin, and hyaluronan, lining
the space of Disse. Activated stellate cells also release proinflammatory, profibrogenic,
and promitotic cytokines. Other features of stellate cells include the ability to
present antigen and their consistent contact with individual fibers of autonomic nerve
endings.
Hepatic blood flow can be significantly modified by activated stellate cells. Blood
flow through sinusoids can be restricted by the contraction of the stellate cells
that extend processes around the sinusoidal endothelial cells. In health, there is
a balance between the nitric oxide–stimulated vasodilation and endothelin-1–driven
vasoconstriction. During active fibrosis and cirrhosis, there is an increase of endothelin-1,
produced by sinusoidal endothelial cells causing contraction of the stellate cell–derived
myofibroblasts.
Fibrosis can form within portal tracts expanding their area, or as septa extending
into the parenchyma and eventually bridging between portal tracts or central veins.
Fibrosis in the portal tracts is primarily generated by the activity of portal fibroblasts,
rather than hepatic stellate cells. Biliary injury leads to a rapid proliferation
of periductular myofibroblasts derived from portal fibroblasts and ECM production.
The volume of the portal tracts is increased by the generation of the ECM, ductular
proliferation, myofibroblast proliferation, and edema.
Fibrosis can be less florid and more insidious when it lines the space of Disse, disrupting
the normal exchange between the plasma and hepatocyte microvilli. The essential microvascular
unit of the liver is the sinusoid, lined by fenestrated endothelial cells, and separated
from the hepatocytes by the space of Disse. A porous basal membrane–like matrix in
the space of Disse helps to maintain the differentiated states of the hepatocytes
and hepatic stellate cells that inhabit the space of Disse. When there is excess ECM
lining the space of Disse, the adjacent sinusoidal endothelial cells lose their characteristic
fenestrations, and the sinusoids are transformed functionally into capillaries. This
process is known as capillarization and leads to significantly impaired exchange between
the hepatocytes and the plasma. This causes diminished hepatic function when the lesion
is extensive. The cells responsible for the ECM deposition are primarily in the myofibroblast
family, including hepatic stellate cells.
Because collagen has a finite half-life, and as hepatic fibrosis exists in a balance
of synthesis and removal, it is potentially reversible. Elimination of the source
of injury or successful therapy can lead to stellate cell apoptosis and a reduction
in the synthesis of inhibitors of the enzymes responsible for the degradation of the
collagen and other ECM elements, primarily matrix metalloproteinases. This has been
demonstrated in rodent models of hepatic fibrosis produced by bile duct ligation,
and models using carbon tetrachloride. However, over time, collagen can mature and
fibrils can cross-link and become resistant to enzymatic degradation, leading to permanent
fibrosis. Long-term or permanent fibrotic changes can also be caused by severe acute
injury that leads to an initial focus of fibrosis and is so severe that it affects
local vascular supply.
The distribution of fibrosis in the liver can reflect the pathogenesis of the injury
responsible.
•
Inflammatory disorders that produce piecemeal necrosis of hepatocytes, typically viral
hepatitis in humans and often an idiopathic disorder in veterinary species, can evolve
to form portal-to-portal, or portal-to-central, fibrous septa.
•
In the event of inflammation or obstruction involving the bile ducts, proliferation
of periductular myofibroblasts and prominent ductular reaction can lead to biliary
fibrosis or a pattern of portal-to-portal bridging termed biliary cirrhosis when accompanied
by nodular regenerative proliferation of hepatocytes (Fig. 2-43
).
Figure 2-43
Bridging portal fibrosis and bile duct hyperplasia secondary to bile duct obstruction
in a horse.
•
Postnecrotic scarring
occurs after massive necrosis, where large areas of parenchyma are destroyed. The
reticulin network collapses and condenses and portal areas converge, resulting in
broad, irregular bands of scar tissue with variable irregular areas of parenchymal
regeneration interspersed (Fig. 2-44
).
Figure 2-44
Postnecrotic scarring in the liver of a sheep.
(Courtesy P. Stromberg)
•
Diffuse hepatic fibrosis is the outcome of chronic parenchymal injury, such as prolonged
inflammation or multiple episodes of zonal necrosis. The fibrosis throughout the lobules
bridges connective-tissue tracts in portal areas and hepatic venules to produce pseudolobulation,
in which small areas of parenchyma are separated by a pattern of fibrosis on the scale
of true lobules. This pattern of fibrosis can lead to capillarization of sinusoids,
or intrahepatic bypass of portal and arterial blood, both of which can reduce the
influences of growth factors and nutrients on liver regeneration. The accompanying
hypoxia is important in the genesis of the hepatocellular atrophy that is almost always
concomitant with diffuse hepatic fibrosis.
•
Centrilobular (periacinar) fibrosis is the most common pattern following zonal necrosis
in hypoxic and toxic injury. Good examples are seen in animals with prolonged passive
venous congestion of the liver, especially when this is caused by extracardiac sources
of increased venous pressure, rather than to congestive heart failure (Fig. 2-45
). Otherwise, this pattern of fibrosis is a response to toxic injury; poisoning by
pyrrolizidine alkaloids may cause it in ruminants, and extraordinary development of
periacinar fibrosis may follow accidental exposure to nitrosamines in several species.
Figure 2-45
Centrilobular fibrosis (cardiac fibrosis), most severe at the center of the lobe in
chronic passive congestion in a dog.
Cirrhosis
Cirrhosis has several definitions, and consensus regarding the essential features
is difficult to obtain. In general, it is agreed that cirrhosis is not merely an end
stage of ECM accumulation in the liver, but, in fact, is a multifaceted distortion
of hepatic parenchymal architecture and hepatic vascular anatomy. By one definition,
cirrhosis is “a diffuse process characterized by fibrosis and the conversion of normal
liver architecture into structurally abnormal nodules.” The key features are the formation
of regenerative nodules of hepatocytes surrounded by fibrous septa and vascular disorders
that often integrate both central veins and portal tracts. Other than dogs, most veterinary
species develop diffuse hepatic fibrosis following chronic injury, but do not develop
regenerative nodules. Chronically injured canine livers often have a robust formation
of regenerative nodules separated by fibrous septa typical of cirrhosis (Fig. 2-46A-C
). Portosystemic shunts and venous occlusion may result, interfering with hepatic
function and driving portal hypertension. Note that not all nodular livers are cirrhotic.
Clinically, cirrhosis results in hepatic insufficiency, mainly in the form of ascites
and hypoproteinemia rather than excretory dysfunction and icterus, although, in advanced
cases, all manifestations of fatal liver failure can occur. The hallmarks of cirrhosis
are:
1.
Bridging fibrous septa, ranging from delicate bands to broad scars that replace multiple
adjacent lobules. These fibrous septa contain vascular channels, originating from
sinusoids in collapsed parenchyma or angiogenesis that allow a considerable portion
of blood to bypass hepatocytes. These microvascular channels in fibrous septa resist
flow more than normal sinusoids. This factor, as well as the relative increase in
flow in these vessels shunting around the nodules, contributes to portal hypertension.
2.
Impaired exchange between hepatocytes and sinusoidal blood, because of the increase
in perisinusoidal ECM.
3.
Parenchymal nodules, created by the regenerative attempts of entrapped hepatocytes,
and varying in size from <3 mm in diameter (micronodular) to several centimeters in
diameter (macronodular). These nodules are composed of trabeculae that are typically
2 or more cells thick, with relative reduction of sinusoidal space. Some expanding
nodules may compress the vessels within fibrous septa, contributing to portal hypertension.
Nodule formation is prominent in dogs, but significantly less so in most other domestic
species.
4.
Ongoing damage and reorganization of the hepatic connective tissue, sometimes with
areas of portal and arterial thrombosis and segmental ischemia is typical, but not
always present.
Figure 2-46
A. Regenerative nodules in the liver of a dog. B. Cut surface of the liver. C. Microscopic
appearance of the regenerative nodule and adjacent septum.
Cirrhosis is usually the end result of several pathogenic processes, namely, cell
death (necrosis or apoptosis) and active inflammation with chronic fibrosis. Cirrhosis
is not a synonym for chronic hepatic fibrosis, although some insults that cause chronic
diffuse fibrosis in the liver can lead to cirrhosis. The nodular pattern of regeneration
in cirrhosis is caused by expanding islands of surviving parenchyma entrapped between
bands of scar tissue. At the point of liver failure, these regenerative attempts are
insufficient to restore function. Despite the increased hepatocellular mass, the inability
to restore normal function is attributed to altered circulation through the bridging
scar tissue, portal hypertension and shunting, deposition of ECM in the perisinusoidal
space, reduced supply of portal growth factors, inhibition of proliferation of ECM,
and toxic inhibition of hepatocellular proliferation. The characteristic fibrovascular
septa that bridge portal and central vascular tracts are readily observed in liver
sections, but the portal-hepatic shunting therein can be difficult to identify. Clinical
evidence for portosystemic shunting, such as impaired ammonia or bile acid clearance,
can reflect acquired portocaval shunts that often develop as a result of portal hypertension
from chronic hepatic fibrosis.
Acquired portosystemic shunting
Any chronic liver disease that causes sufficient fibrosis or atrophy to restrict portal
blood flow significantly and produce portal hypertension has the potential to cause
the development of collateral portosystemic shunts. These are described later in the
section on Vascular factors in hepatic injury and circulatory disorders.
Further reading
Anthony PP, et al. The morphology of cirrhosis. Recommendations on definition, nomenclature,
and classification by a working group sponsored by the World Health Organization.
J Clin Pathol 1978;31:395-414.
Desmet VJ. Ductal plates in hepatic ductular reactions. Hypothesis and implications.
I. Types of ductular reaction reconsidered. Virchows Arch 2011;458:251-259.
Desmet VJ. Ductal plates in hepatic ductular reactions. Hypothesis and implications.
III. Implications for liver pathology. Virchows Arch 2011;458:271-279.
Dranoff JA, Wells RG. Portal fibroblasts: underappreciated mediators of biliary fibrosis.
Hepatol 2010;51:1438-1444.
Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annu Rev Pathol 2011;6:425-456.
Michalopoulos GK, DeFrances MC. Liver regeneration. Science 1997;276:60-66.
Overturf K, et al. Serial transplantation reveals the stem-cell-like regenerative
potential of adult mouse hepatocytes. Am J Pathol 1997;151:1273-1280.
Hepatic Dysfunction
Hepatic failure is a syndrome that results from inadequate hepatic function. The liver
provides numerous essential processes, and liver failure can affect metabolic, synthetic,
catabolic (detoxification), and immune functions. There is no specific point at which
diminished function can clearly be defined as “failure,” given the various functions
of the liver, but it is best understood as the point at which liver function is not
capable of sustaining life. The liver is an organ with very large functional capacity
and regenerative potential. Signs of insufficiency do not develop until the reserves
in production, supply, and recovery are exhausted. The central role of the liver in
many systemic functions means that liver disease can be manifested as lesions or dysfunctions
elsewhere, for example, in the brain, skin, blood, and gut. Failure may develop suddenly,
frequently following exposure to toxic compounds, or failure may develop in a slow
inexorable fashion as a chronic disease processes. Hepatic failure, particularly chronic
liver failure, may not affect all processes equally, and one or more features of liver
failure may predominate in individual cases.
Acute liver failure is uncommon, and is clinically evidenced as a severe and rapid
liver injury. There are a number of causes. The consequences are also multiple and
varied, including abrupt loss of normal metabolic and immunologic functions, as well
as hepatic encephalopathy, coagulopathy, jaundice, photosensitization (in herbivores),
and possibly dysfunction of other organs. In humans, there are recognized viral infections
that can cause acute liver failure, but there are currently no known animal viruses
capable of causing this syndrome, although recent studies suggest that equine serum
hepatitis (Theiler's disease) may be caused by an equine pegivirus. Toxin-induced
acute liver failure is likely the most common form in veterinary medicine. The toxic
injury may come from natural sources, such as those found in plants and fungi, or
from overdose of or idiosyncratic reactions to therapeutic drugs. Hyperthermia from
environmental heating or prolonged seizure activity can also cause acute hepatic failure.
Acute ischemic injury from profound hypotension secondary to sepsis or cardiac failure
may also occur.
Chronic liver failure results from progressive destructive processes, such as fibrosis
or inflammation, or a combination of the two. The hallmarks of chronic liver failure
in dogs are fibrosis and nodular regeneration, but other species are more prone to
a fibrotic response alone. The progressive loss of critical functional hepatic mass
caused by ongoing hepatocyte injury, shunting of portal blood within fibrous septa
bypassing hepatocytes, and abnormal bile drainage, lead to a terminal stage of hepatic
insufficiency.
Chronic liver failure can be subdivided into 2 forms: end-stage liver, the typical
fibrotic liver with progressive loss of functional hepatic mass, and “acute on chronic
failure,” a circumstance in which a patient with compensated chronic liver disease
acutely develops failure. The causes for the acute transition to failure are not certain,
but the buildup of various toxins, such as aromatic amino acids, benzodiazepines,
and ammonia, among others, may be involved. Other theories include increased bacterial
or fungal translocation from the gut, leading to hepatic infection and sepsis.
Liver failure, whether acute or chronic, affects a number of critical processes. These
disturbances include hepatic encephalopathy, cholestasis and jaundice, hepatogenous
photosensitization, hemorrhagic diathesis, hepatorenal syndrome, ascites, and hepatocutaneous
syndrome.
Hepatic encephalopathy (HE)
The neurologic manifestations of hepatic failure are variable and nonspecific; they
range from dullness, through complete unawareness and compulsive aimless movement,
to mania and generalized convulsions. There is considerable variation in the clinical
signs of hepatic encephalopathy between different species. Sheep rarely show more
than dullness and central blindness, with perhaps some compulsive chewing movements
and tremor. The picture in cattle is similar, but mania and aggression may also be
seen, whereas frenzy is more often recorded in horses. The closer clinical observation
of cats and dogs may reveal more subtle behavioral changes, and inappetence and vomiting
are commonly reported in carnivores with portosystemic shunts. Ptyalism is common
in cats with HE.
The clinical context in which HE occurs can be important. In animals with acute hepatic
disease, HE is a serious sign, usually indicating imminent death. However, in animals
with more chronic conditions, such as portosystemic shunting or deficiency of a urea
cycle enzyme, neurologic signs can be intermittent for many months, and may disappear
after appropriate dietary modification. The pathogenesis of HE involves more than
just neurons, as astrocyte and glial cell responses are now evident. Older literature,
particularly in vitro studies, which did not take all of these features into account,
will likely need reinterpretation.
Ammonia toxicity is regarded as the major part of the clinical signs and the brain
lesions; however, there are a number of other factors that may play a role. Blood
ammonia is largely of dietary origin, derived from protein and urea by microflora
in the large bowel. Ammonia is also derived from hepatic deamination of amino acids
and from metabolism of glutamine in peripheral tissues. Ammonia is normally removed
in the first pass of portal blood through the liver, wherein it is incorporated with
carbon dioxide into carbamoyl phosphate that enters the urea cycle. In shunting or
hepatic failure, ammonia that bypasses hepatic clearance accumulates in the circulation.
Ammonia is able to cross the blood-brain barrier, and initially, this influx leads
to astrocyte injury. Astrocytes are the site of ammonia detoxification in the brain,
and they eliminate ammonia by the synthesis of glutamine through amidation of glutamate
by the enzyme glutamine synthetase. Elevated blood ammonia increases the accumulation
of glutamine in astrocytes, resulting in osmotic stress and astrocyte swelling caused
by cytotoxic edema. Affected astrocytes that are adjacent to endothelium cannot maintain
the blood-brain barrier, and this permits increased ammonia entry into the brain.
Direct ammonia-induced effects on brain endothelial cells are also possible. Briefly,
the consequences of increased ammonia in the brain include a net decrease in energy
metabolism within the brain, increased edema formation because of astrocyte dysfunction,
and injury to neurons, leading to an imbalance in neurotransmitters that emphasizes
neural inhibition.
The pathogenesis of HE may vary depending on the type of liver failure—acute or chronic—that
produces the cerebral injury. Also, not all parts of the brain are affected in a similar
fashion. In acute hepatic failure, cerebral edema may be the leading cause of death
because of increased intracerebral pressure with possible herniation. The increase
in intracranial pressure is, however, linked to brain ammonia levels. Currently, it
is thought that swollen astrocytes release vasogenic factors, such as nitric oxide,
causing intracerebral hyperemia, and that this leads to edema formation. Systemic
inflammatory responses can enhance the severity of HE. Neuroinflammation, mediated
by microglia, is evident in acute liver failure and tends to increase with increasing
duration of HE. Synergetic interactions between increased ammonia and systemic or
neuroinflammation can increase the severity of HE. However, the specific mechanisms
by which systemic inflammation triggers neuroinflammation are not currently known.
In chronic hepatic failure, ammonia and neuroinflammation are likely to contribute
synergistically to HE. Ammonia plays a role as a directly neurotoxic agent, altering
neurotransmission, and potentially contributing to cerebral energy failure through
inhibition of α-ketoglutarate dehydrogenase, a rate-limiting enzyme in the tricarboxylic
acid cycle. Chronic increase in brain ammonia is associated with disrupted neural
transmission involving all of the different neurotransmitter systems of the brain.
The main neurotransmitter systems affected involve neuropeptides, with an evolution
toward an increase in the inhibitory γ-aminobutyric acid (GABA)ergic system, and downregulation
of the excitatory glutaminergic system. Increased intracerebral ammonia leads to an
increase in extracellular glutamate, which will initially drive upregulation of N-methyl-D-aspartate
(NMDA) receptors, causing neuronal damage, but eventually, there is a reduction in
NMDA receptors and glutaminergic signaling, leading to neuroinhibition. In addition,
increased ammonia alone has been linked to neuroinflammation, possibly through direct
activation of microglia, which also interferes with normal neural transmission.
Newer studies in animal models have demonstrated that agents acting on specific targets
in the brain, including phosphodiesterase 5, type A GABA receptors, and mitogen-activated
kinase (p38), can improve cognitive function in mild forms of experimental hepatic
encephalopathy. Survival can be increased by treatment with NMDA receptor agonists.
Studies have also implicated alterations in inhibitory and excitatory neurotransmitters,
including endogenous benzodiazepines, and serotonin, as well as their receptors. Although
infusion of ammonia or hyperammonemia caused by deficiency of urea cycle enzymes reproduces
similar neurologic signs and vacuolar lesions, other noxious substances in the alimentary
tract are also believed to contribute to hepatic encephalopathy resulting from liver
failure. These include variably toxic amines, captans, thiols, and short-chain fatty
acids, which are normally removed from the portal blood in one passage through the
liver after production in the large bowel. Thus there are many factors to consider
in the pathogenesis of HE.
The microscopic lesions of HE are subtle and variable, likely because postmortem examinations
of the brain are performed at various time points of chronicity and clinical severity.
There are also significant differences in the microscopic lesions between species.
In humans, the hallmark of HE is Alzheimer type II astrocytosis, in which astrocytes
are enlarged with swollen nuclei, margination of chromatin, and prominent nucleoli.
These can be found in large animals, particularly horses. In dogs, Alzheimer type
II changes are rare if they occur at all, but vacuoles in gray matter, particularly
in brain stem nuclei, predominate. Lesions in cats have features of both the dog and
the horse (see Vol. 1, Nervous system). These changes can be observed best in animals
with chronic liver disease and portosystemic shunts; the brain lesions can be minimal
in animals with acute liver failure.
Cholestasis and jaundice
Cholestasis is the term for impaired bile secretion and flow as well as a failure
to secrete organic and inorganic components of bile, with accumulation of these elements
in blood. Normal bile formation and flow is dependent on the activity of a series
of membrane transporters found in enterocytes, hepatocytes, and biliary epithelium.
In rare cases, hereditary mutations can lead to cholestasis, but more often, cholestasis
is caused by exposure to injurious drugs, hormones, proinflammatory cytokines, or
obstruction. Clinically, cholestasis leads to increased levels of bilirubin and bile
acids in the blood because of retention. Injury to the biliary tree leads to increases
in serum alkaline phosphatase. Disturbance of bile flow can originate from altered
function of hepatocytes, termed hepatocellular cholestasis, or because of obstruction
of the biliary tree, termed obstructive cholestasis. Hepatocellular cholestasis can
be attributed to impaired uptake, metabolism, secretion, or transport of bile constituents.
Obstructive cholestasis is related to obstruction of bile flow at the level of the
major bile ducts or gallbladder.
Jaundice (icterus) occurs when the tissues, particularly the sclerae, are pigmented
yellow because of an excess of bile pigments, primarily bilirubin, in the plasma.
Jaundice can arise from hepatocellular cholestasis or obstructive cholestasis and,
in addition, from prehepatic causes, such as an overproduction of bilirubin from heme
catabolism in hemolytic diseases (Fig. 2-47
). The associated hypoxia caused by anemia may facilitate cholestasis in hemolytic
diseases.
Figure 2-47
Deeply bile-stained liver from a jaundiced dog with immune hemolytic anemia and cholecystitis.
(Courtesy A.P. Loretti.)
Histologically, both forms of cholestasis share common features, but there are additional
lesions associated with duct obstruction in the portal tracts and bile ducts. The
main histologic manifestation of cholestasis is accumulation of homogeneous waxy brown
bile pigment in bile canaliculi. Bile regurgitated from hepatocytes can also be found
in Kupffer cells following phagocytosis. Bilirubin can be found in the hepatocellular
cytoplasm in most species, especially if frozen sections are examined, but it is quite
rare for bile to be evident in canine hepatocytes. Bile accumulation is more severe
in the centrilobular regions of the lobules, but can extend to the periportal regions
in severe cases. With time, canalicular plugs are cleared, and they may not be evident
in more chronic cases of cholestasis. Hepatocyte rosettes, collections of 2 or more
hepatocytes surrounding a dilated canaliculus are common. Hepatocellular degeneration
may also be evident, sometimes with bile staining of the cytoplasm of injured hepatocytes.
Scattered apoptotic cells may be present, although significant necrosis is not usually
a feature of cholestasis. In more severe cases of obstructive cholestasis, confluent
areas of bile-stained hepatocellular necrosis, termed bile infarcts, can develop.
Bile infarcts are rare in dogs and cats, but can be found in other species. Cholate
stasis refers to hepatocellular degeneration in the periportal region, often accompanied
by a proliferation of small-caliber bile ducts (ductular reaction). Affected hepatocytes
have swollen pale cytoplasm, and they may contain copper granules. This lesion is
most common in horses with obstruction and rare in dogs. In hepatocellular cholestasis,
there are no distinctive lesions in the portal tracts. However, in obstructive cholestasis,
there are a number of distinctive changes. Initially, there is edema in the portal
tract and an inflammatory infiltrate that is most often predominantly neutrophilic.
There is an accompanying proliferation of small-caliber bile ducts arranged in an
apparently haphazard fashion (ductular reaction). There may be degeneration or proliferative
changes in pre-existing interlobular bile ducts, often associated with inflammation,
depending on the cause of the obstruction. Ductal ectasia may also be present. With
time, there is an increase of fibrosis that expands the portal tract outline and can
bridge between portal tracts in cases of severe obstruction. In addition, there is
concentric fibrosis surrounding the bile ducts. Proliferation of bile ducts, termed
ductular reaction, is a constant feature. Mixed inflammatory cells, usually with fewer
neutrophils than that seen in acute cholestasis, as well as macrophages containing
pigment found in the portal tract.
Hepatocellular cholestasis arises when there is a disturbance in 1 or more of the
3 main steps involved in bile excretion. They include failure (1) to take up bilirubin,
bile acids, or other bile constituents; (2) to conjugate bilirubin, bile acids, or
other constituents; and (3) to transport and excrete conjugated bilirubin, bile acids,
or other bile constituents into canaliculi. Most of these bile metabolism issues are
mediated by a series of hepatobiliary transporters, molecular pumps involved in the
transport of bilirubin, bile acids, and other organic ions in hepatocytes, canaliculi,
and biliary epithelium, although some pumps serve to export substances as well. Hepatocellular
uptake of unconjugated and conjugated bilirubin, like other organic ions, is mediated
by members of the organic anion-transporting polypeptide (OATP) family found on the
basolateral aspects of hepatocytes. Bilirubin is conjugated by uridine diphosphate–glucuronyl
transferase in the endoplasmic reticulum of hepatocytes. From there it is transported
into the bile. Bile acids are imported from the plasma by the Na+-taurocholate cotransporting
polypeptide (NTCP). Transport of bile acids, the main osmotic driving force for bile
formation (bile acid–dependent flow), into the canaliculus is performed by the bile
salt export pump (BSEP). Aquaporin channels facilitate the movement of water into
the canaliculus in response to the osmotic forces produced by the bile acids. Movement
of bilirubin into the canaliculus for excretion is accomplished by the multidrug-resistance–associated
protein-2 (MRP-2). Reduced glutathione is also a substrate for MRP-2, and it is reduced
glutathione and bicarbonate that are the main elements of the bile acid–independent
portion of bile flow, the second most significant force in bile flow. Bile is continually
modified by transporter-driven processes of secretion and absorption during its transit
to the duodenum.
In hepatocellular cholestasis, drugs or endotoxin-induced proinflammatory cytokines
interfere with the activities of molecular pumps or inhibit their synthesis. Endotoxin
exposure both reduces activity of OATP and NCTP, but also interferes with the activity
of BSEP and MRP-2.
Hepatocellular cholestasis can also occur through disruption of the structural integrity
of the canaliculi, as discussed later for Lantana camara toxicity. Poisons such as
phalloidin and cytochalasin, which disrupt the polymerization cycle of pericanalicular
actin microfilaments, are cholestatic because these contractile filaments are required
for propelling bile along the canaliculi. In horses, fasting can cause plasma bilirubin
increases and jaundice. The mechanism may be the impaired uptake of bilirubin from
the plasma or possibly impaired conjugation of bilirubin when energy supplies are
low. Rarely, cholestasis and its indicators can occur in the absence of significant
hepatocellular damage and other signs of liver failure. This can occur in toxicity
by L. camara in ruminants and as an idiosyncratic reaction to some drugs.
In obstructive cholestasis, the activities of NTCP and OATP transporters are both
reduced, causing bile acids and bilirubin to accumulate in the blood. Transport of
bile acids out of the hepatocyte and into the canaliculus is maintained by BSEP. The
regulation of transcription of the various transporters in hepatocellular or obstructive
cholestasis is controlled by the interaction of bilirubin or bile acids with several
nuclear receptors, primarily PXR (pregnane X receptor) and, to a lesser extent, others,
such as CAR (constitutive androstane receptor) and FXR (farnesoid X receptor). During
cholestasis, nuclear receptors orchestrate protection of hepatocytes from injury by
reducing the expression of basolateral uptake transporters and increasing the expression
of basolateral export pumps, as well as reducing new bile acid synthesis and increasing
the rate of intrahepatic metabolism of bile acids. However, these adaptive responses
are not always sufficient to prevent cholate-induced hepatotoxicity.
Severe diffuse liver necrosis obviously impairs bile excretion at various levels,
but the severity of cholestasis or of jaundice depends on the amount of liver affected,
the chronicity, the supply of heme for catabolism, and nonhepatic routes of bilirubin
excretion. Focal liver injury can have substantial local cholestasis, but clinically
detectable cholestasis does not occur because bilirubin can be cleared by unaffected
parts of the liver. Similarly, segmental duct obstructions that spare some parts of
the liver can lead to cholestasis but not jaundice. This can occur in anorexic or
dehydrated cats, in which bile may become dehydrated and viscous, causing ductal obstruction.
Some of the various transporters and conjugating enzymes involved in bilirubin excretion
can be genetically defective. Congenital hyperbilirubinemia in mutant Southdown sheep
is the result of impaired hepatic uptake of unconjugated bilirubin. These animals
have few liver lesions, but eventually develop chronic renal disease, the reason for
which is not clear. Unconjugated bilirubin levels in the plasma are consistently elevated,
but sufficient excretion takes place to prevent them from becoming icteric. They become
photosensitized, indicating that excretion of phylloerythrin (phytoporphyrin) is less
efficient than that of bilirubin.
Hyperbilirubinemia in mutant Corriedale sheep is a defect in excretion of conjugated
bilirubin similar to Dubin-Johnson syndrome in humans and mutant rat strains. In humans
and rats, there is a mutation in Mrp-2 causing hypofunction and a similar mutation
is likely in affected sheep. Affected sheep have an elevation of plasma bilirubin
(just over half of which is conjugated), but there is no obvious jaundice. Nevertheless,
phylloerythrin (phytoporphyrin) excretion in these Corriedale sheep is also sufficiently
impaired to produce photosensitization. There is impaired excretion of other conjugated
metabolites, and there is dark pigmentation of the liver by polymerized residues of
retained catecholamine metabolites. This pigment, resembling lipofuscin, accumulates
in lysosomes in the pericanalicular cytoplasm.
The recognition of jaundice at postmortem sometimes involves differentiation of bile
staining of tissues from the yellow staining caused by accumulation of carotenoid
pigments. These latter are limited to fat depots and are to be expected in certain
species such as horses; sometimes there are breed influences, as seen in the yellow
fat of Channel Island breeds of dairy cattle. The yellow discoloration of fat depots
of older cats is less well understood. In animals fed ox liver, carotenoids may again
be responsible, and in others, there may be some contribution by ceroid-type pigments.
Distinction of the fatty pigments from bile depends on the absence of the former from
pale, nonfatty tissues, such as periosteum and dermal collagen.
Photosensitization
Photosensitization is the term applied to inflammation of skin (usually unpigmented)
because of the action of ultraviolet light of wavelengths 290-400 nm on photodynamic
compounds that have become bound to dermal cells. In primary photosensitization, these
compounds may have been deposited unchanged in the skin after ingestion, before the
normal liver is capable of excreting the native compound. This is seen, for example,
after ingestion of hypericin in St. John's wort (Hypericum perforatum). Photodynamic
agents may also be produced by aberrant endogenous metabolism. This can occur, for
example, in congenital erythropoietic protoporphyria caused by ferrochelatase deficiency
in Limousin and Blonde d'Aquitaine calves.
Hepatogenous photosensitization almost always accompanies cholestasis of more than
a few days' duration in herbivores that are kept in sunlight and that have been eating
green feed. Phytoporphyrins (formerly termed phylloerythrins) are green photoactive
catabolites of plant porphyrins (mainly chlorophyll) that are generated by the alimentary
microflora of herbivores. Some phytoporphyrin is absorbed and normally excreted in
the bile by the transporters that eliminate bilirubin. Cholestasis in herbivores can
increase retention of phytoporphyrin in the blood, and it can result in photosensitive
dermatitis of unpigmented areas of skin exposed to sunlight for several days (Fig.
2-48
). It is possible, however, for mild photosensitization to appear in the absence of
gross or microscopic evidence of cholestasis in animals grazing alfalfa, Paspalum,
pangola, or Panicum grasses; however, it is unclear if these are related to phytoporphyrin
or other photoactive products of these forages. Absence of serum biochemical evidence
for cholestasis or liver damage is used to differentiate primary from secondary photosensitization
in these circumstances.
Figure 2-48
Hepatogenous photosensitization caused by Panicum miliaceum (French millet) in a sheep.
(Courtesy P. Hoskin.)
If no hepatic changes can be discerned in photosensitized animals, the possibility
of primary photosensitization must be considered, but hepatogenous photosensitization
cannot be excluded unless adequate liver function tests are performed.
Hemorrhage and liver failure
Hemorrhagic diathesis characterized by widespread ecchymoses and petechiae can occur
when the liver is injured and becomes the site of significant clotting factor consumption.
However, recent investigations suggest that clinically significant hemorrhage is not
common in acute liver failure. The liver is the source of plasma proteins involved
in the clotting cascade, but these are normally supplied in substantial excess, and
can be induced as part of the acute-phase response to inflammation. Thus coagulopathy
with hemorrhage is most likely to occur when there is acute liver necrosis, for example,
in acute canine infectious hepatitis or xylitol toxicity. Under these conditions,
there is significant intrahepatic consumption of clotting factors at sites of endothelial
necrosis in the damaged liver. Although the damaged liver also fails to resupply the
clotting factors, consumption is probably a minor contributing influence. In more
chronic liver diseases with hypoproteinemia, coagulation tests may be prolonged, but
hemorrhagic diathesis is unlikely, unless there is an additional demand for hemostasis,
for example, during the trauma of surgery. Although production of clotting factors
is often reduced in liver failure, there is a commensurate reduction in anticoagulant
synthesis as well. Portal hypertension and endothelial cell dysfunction may also be
a contributing cause for bleeding in chronic liver disease. Overall, the pathogenesis
of hemorrhagic tendencies in chronic liver disease is complex, multifactorial, and
incompletely understood. Consumption of clotting factors can also occur in septic
diseases that affect the liver and other tissues.
Nephropathy
Acute liver failure may be accompanied by oliguria and biochemical indications of
renal failure. Hepatorenal syndrome is a complication of advanced cirrhosis in humans,
characterized by renal failure in which there are no intrinsic renal morphologic or
functional causes. The pathogenesis of hepatorenal syndrome is related to deterioration
in effective arterial blood volume because of splanchnic arterial vasodilation and
reduced venous return and cardiac output. Intense compensatory vasoconstriction of
the renal circulation results in decreased glomerular filtration and resultant renal
failure. In addition, some hepatic toxins such as acetaminophen may produce injury
in the nephrons as well.
Edema and ascites
Ascites (retention of excess low-protein peritoneal fluid) is a feature of chronic
liver failure, but is more frequently associated with systemic venous congestion (e.g.,
right-sided heart failure) or hypoproteinemia secondary to protein-losing renal or
alimentary tract conditions. It is likely that ascites in advanced liver disease is
influenced by both mechanical and dynamic influences on blood flow through a damaged
liver. Reduced synthesis of albumin and globulins by the failing liver can reduce
vascular oncotic pressure, but edema resulting from this mechanism is generalized.
Experimentally, restriction of blood flow from the liver leads to a swift increase
in sinusoidal plasma within the space of Disse, leading to increased lymph flow into
the thoracic duct and through the hepatic capsule. This explains rapid ascites formation
in cases of hepatic outflow obstruction or veno-occlusive diseases.
There are several theories that explain the complicated process of ascites formation
in chronic liver disease. The theory that best encompasses the known circulatory changes
is known as the peripheral arteriolar vasodilation hypothesis. In end-stage livers,
there is progressive sinusoidal and portal vein hypertension with collateral vein
formation and acquisition of shunting vessels to the systemic vasculature. Sinusoidal
hypertension appears to be an important feature as ascites rarely develops with prehepatic
portal hypertension. The main factor leading to ascites formation is splanchnic vasodilation.
In chronic liver failure, there is increasing portal vein hypertension and a local
release of vasodilators, such as nitric oxide, leading to splanchnic arterial dilation.
With progression, the extent of the arterial vasodilation increases to the point that
effective arterial volume and pressure drops, leading to activation of vasoconstrictor
and antinatriuretic factors, including activation of the renin-angiotensin-aldosterone
system and the sympathetic nervous system, promoting retention of sodium and fluid.
Intestinal capillary pressure and permeability are increased as a result of the increased
portal vein hypertension, promoting excess fluid transit to the abdominal cavity.
Over time, sodium retention by the kidneys is insufficient to compensate for the progressive
arteriolar vasodilation as well as the movement of sodium and fluid to the extracellular
space of the abdomen. This movement is enhanced by hypoproteinemia caused by reduced
synthesis by the liver and dilution secondary to fluid retention. Consequently, there
is persistent activation of antinatriuretic systems, and fluid continues to accumulate.
There is evidence that increased venous return caused by reduced peripheral arterial
vasodilation stimulates endogenous natriuretic substances, but this response is insufficient
to counteract the more significant release of antinatriuretic signals.
Retention of peritoneal fluid can also sometimes result from mechanical obstructions
to mesenteric and peritoneal lymphatics by inflammatory or neoplastic lesions. Peritoneal
fluids with a higher protein and cell content that accumulate in various abdominal
inflammatory conditions are considered exudates (see section on Peritonitis in Vol.
2, Alimentary system and peritoneum).
Hepatocutaneous syndrome
An idiopathic vacuolar hepatopathy with parenchymal collapse and nodular regeneration
has been reported in dogs and cats that have been presented clinically with hepatocutaneous
syndrome (necrolytic migratory erythema, superficial necrolytic dermatitis).
The skin disease resembles necrolytic migratory erythema in humans, a well-defined
paraneoplastic syndrome typically associated with hyperglucagonemia secondary to glucagon-secreting
pancreatic neoplasia, but also reported in individuals with hepatitis, cirrhosis,
celiac disease, chronic malabsorption, and inflammatory bowel disease. In dogs, hepatocutaneous
syndrome is most commonly associated with liver disease, including severe vacuolar
hepatopathy, idiopathic hepatocellular collapse, and hepatopathy secondary to anticonvulsant
drug administration, and more rarely with glucagonoma and gastric carcinoma.
Clinical presentation is typically because of the dermatitis; skin lesions include
erythema, crusting, exudation, ulceration, and alopecia, affecting footpads, periocular,
perioral, anogenital regions, and pressure points. The dermatologic lesions are described
more fully in Vol. 1, Integumentary system. Affected dogs have depressed plasma amino
acid concentrations, inconsistent elevations of plasma glucagon levels, and may also
become diabetic. The liver of affected dogs is usually grossly nodular, resembling
cirrhosis (eFig. 2-10A); however, histologically, there is typically moderate to severe
vacuolation of hepatocytes, with parenchymal collapse accompanied by nodular regeneration
(eFig. 2-10B). Inflammation, necrosis, and fibrosis are not usually prominent. Although
some studies report fibrosis typical of cirrhosis, more characteristically, there
is a network of reticulin and fine collagen fibers representing the remnants of collapsed
hepatic lobules, with proliferation of bile ductules. The vacuolated hepatocytes stain
with oil red O for lipids. The hepatic lesions have been suggested to support an underlying
metabolic, hormonal, or toxic etiology. In humans, persistent hyperglucagonemia stimulates
prolonged gluconeogenesis, resulting in secondary hypoaminoacidemia. Although hepatic
insufficiency can increase glucagon levels in animal models, in dogs, the link between
liver disease and hypoaminoacidemia remains unclear. A hypermetabolic state with exaggerated
amino acid catabolism has, however, been suggested. Other biologically active molecules
that are normally cleared by the normal liver or generated by a damaged liver should
also be considered.
eFigure 2-10
A. Nodular liver from a dog with hepatocutaneous syndrome.
(Courtesy J.L. Caswell.)
B. Hepatic parenchymal collapse with nodular regeneration, hepatocutaneous syndrome.
Further reading
Asakawa MG, et al. Necrolytic migratory erythema associated with a glucagon-producing
primary hepatic neuroendocrine carcinoma in a cat. Vet Dermatol 2013;24:466-469.
Bernal W, et al. Acute liver failure. Lancet 2010;376:190-201.
Gines P, et al. Management of cirrhosis and ascites. N Engl J Med 2004;350:1646-1654.
Gross TL, et al. Superficial necrolytic dermatitis (necrolytic migratory erythema)
in dogs. Vet Pathol 1993;30:75-81.
Kimmel SE, et al. Clinicopathological, ultrasonographic, and histopathological findings
of superficial necrolytic dermatitis with hepatopathy in a cat. J Am Anim Hosp Assoc
2003;39:23-27.
March PA, et al. Superficial necrolytic dermatitis in 11 dogs with a history of phenobarbital
administration (1995-2002). J Vet Intern Med 2004;18:65-74.
Mia AS, et al. Unconjugated bilirubin transport in normal and mutant corriedale sheep
with Dubin-Johnson syndrome. Proc Soc Exp Biol Med 1970;135:33-37.
Mullen KD, Prakash RK. Hepatic Encephalopathy. New York: Humana Press; 2012.
Nyland TG, et al. Hepatic ultrasonographic and pathological findings in dogs with
canine superficial necrolytic dermatitis. Vet Radiol Ultrasound 1996;37:200-205.
Quinn JC, et al. Secondary plant products causing photosensitization in grazing herbivores:
their structure, activity and regulation. Int J Mol Sci 2014;15:1441-1465.
Schrier RW, et al. Peripheral arterial vasodilation hypothesis: a proposal for the
initiation of renal sodium and water retention in cirrhosis. Hepatol 1988;8:1151-1157.
Stravitz RT, et al. Minimal effects of acute liver injury/acute liver failure on hemostasis
as assessed by thromboelastography. J Hepatol 2012;56:129-136.
Tripodi A, Mannucci PM. The coagulopathy of chronic liver disease. N Engl J Med 2011;365:147-156.
Wagner M, et al. New molecular insights into the mechanisms of cholestasis. J Hepatol
2009;51:565-580.
Wagner M, et al. Nuclear receptors in liver disease. Hepatol 2011;53:1023-1034.
Witte ST, Curry SL. Hepatogenous photosensitization in cattle fed a grass hay. J Vet
Diagn Invest 1993;5:133-136.
Postmortem and Agonal Changes in Liver
The liver, rich in nutrients for bacteria and freely exposed to agonal invaders from
the intestine, undergoes postmortem decomposition very rapidly. Gas bubbles generated
by anaerobic saprophytes form first in the hepatic blood vessels, but soon suffuse
large portions of the organ. The vessels and adjacent parenchyma are stained by hemoglobin.
The substance of the organ becomes soft and clay-like, and the formation of putrefactive
gases may make it foamy. On the capsular surface, irregular, pale foci are visible;
they superficially may resemble infarcts or fatty areas but can be observed to increase
in size during the postmortem interval, and microscopically are without cellular reaction.
Bacilli are present in large numbers in such foci. Green-black pigmentation of the
capsule and superficial parenchyma occurs where the liver is in contact with gut,
and the lobes surrounding the gallbladder become stained by bile.
Microscopic structural changes occur in the liver, approaching and immediately following
death. Shrinkage of liver cells, possibly because of a period of anaerobic catabolism
of glycogen, and widening of centrilobular (periacinar) sinusoids because of hepatic
congestion, are seen after death. Dissociation of liver cells may be complete, with
every cell in every cord separated and free from adjacent cells, so architectural
patterns are lost. The early expression of this change affects centrilobular cells,
which become detached, rounded in contour, condensed, and hyperchromatic. The dissociation
is particularly evident in feline panleukopenia and leptospirosis, related in part
to antemortem changes.
Vascular Factors in Hepatic Injury and Circulatory Disorders
Hepatic artery
The hepatic artery delivers approximately of the afferent hepatic blood supply, but
~40-50% of the oxygen. Hepatic artery flow responds to alterations in portal vein
flow to sustain overall perfusion of the liver at a nearly consistent level via what
is termed the hepatic arterial buffer response. Hepatic arterial flow is believed
to be regulated via adenosine levels in the space of Mall. Adenosine is produced at
a constant level, but with decreased portal flow less adenosine is washed out, raising
local concentrations and causing the hepatic artery to dilate and increase arterial
flow. Other mediators of arterial flow are recognized, and they include hydrogen sulfide,
nitric oxide, and the autonomic nervous system.
Complete loss of arterial flow can be, after a brief period of injury, compensated
for via the portal flow because the liver normally only extracts ~40% of the oxygen
supplied by the portal vein. Within the liver, branches of the hepatic artery form
several patterns, including (1) a peribiliary plexus, (2) a vasa vasorum for the portal
vein, or (3) an array of terminal hepatic arterioles that drain directly into the
sinusoids. In addition, the hepatic artery provides branches to supply the liver capsule.
The peribiliary plexus surrounds intrahepatic bile ducts and likely plays a role in
an exchange of bile constituents and vasoactive factors. Hepatic arterial occlusions
occur rather commonly in animals but usually involve small intrahepatic branches and
are of little consequence. Large segments of the liver may be necrotic in cats as
a result of thrombosis of the aorta and hepatic artery. Verminous arteritis may occlude
the hepatic artery in horses. The extent of necrosis depends on how completely the
obstruction excludes collateral circulation and also on the oxygen tension of the
portal blood. Ischemic areas of liver can be sequestered, but in some instances, bacteria
such as clostridia and other anaerobes can flourish and release potent toxins with
systemic effects.
Portal vein
The portal vein drains the large and small intestine, stomach, pancreas, gallbladder,
and spleen and normally contributes of hepatic blood flow. Because the portal vein
collects blood from the abdominal viscera, the rate of flow is variable, depending
on physiologic factors such as eating, which increases, or stress, which decreases,
portal flow. Portal venous blood has considerably more oxygen than most systemic venous
blood and contributes ~60% of the liver's oxygen needs. The portal vein branches successively
until the finest branches, the terminal portal venules, drain into the sinusoids via
side branches, the inlet venules. Entry of blood into the sinusoids is regulated via
sphincters in the terminal inlet venules. Portal blood flow is likely streamlined,
rather than turbulent, but there is little agreement regarding existence of specific
patterns of flow. Streamlining may account for the different regional distributions
sometimes observed with metastatic tumors and infections. The umbilical vein usually
drains to the left lobe, so hematogenous umbilical infections tend to localize in
the left lobe.
The liver cannot regulate portal venous flow, so hepatic blood flow is largely balanced
by arterial supply that varies with the portal venous supply to maintain a relatively
consistent hepatic perfusion. Hepatic microcirculation is well regulated at the level
of inflow by sphincters in the finest branches of the portal venules, the inlet venules,
as well as the terminal branches of the hepatic arterioles and at the level of outflow
by sphincters that regulate passage of blood from the sinusoids to the terminal hepatic
venules. In dogs, the spiral smooth muscle that invests the wall of sublobular hepatic
veins can contract and alter hepatic venous outflow in various conditions, particularly
in shock, where contraction leads to acute congestion and pooling of blood in the
liver and the organs that drain into the portal vein. Hepatic blood thus flows evenly
through the sinusoids under a very-low-pressure gradient. The liver receives ~25%
of the cardiac output, even though it represents only ~2.5% of body mass. Approximately
25% of the weight of the liver in situ is blood.
Obstruction of the portal vein, if sudden and complete, can produce a condition akin
to strangulation of the gut, and death occurs quickly without significant hepatic
change; however, surgical diversion of the portal vein into the vena cava produces
only transient injury to the liver. Obstruction of a large branch of the portal vein
in cattle, sheep, and cats leads to acute ischemia of a wedge of tissue in which necrosis
may be zonal or massive. Obstruction of many small portal radicles is common, with
necrosis of many lobules. It is evident that if a collateral supply develops and oxygenation
remains adequate, obstruction of portal radicles will have no immediate effect on
the hepatic parenchyma, save perhaps to make it more sensitive to toxic injury. The
parenchyma in the affected lobe, deprived of hepatotrophic factors, loses much of
its regenerative power and atrophies fairly rapidly, allowing condensation and scarification
of the stromal tissues. Loss of portal venous inflow and obstruction of hepatic venous
outflow, a double-hit insult, can lead to complete infarction of the liver as seen
in hepatic lobe torsion. Arterial supply can be obstructed as well in this situation.
Acute increase in pressure in the portal vein may occur in any severe episode of widespread
acute hepatic necrosis; the cause appears to be simple obstruction of the sinusoidal
flow by thrombosis and actual sinusoidal disruption. In such animals, there is severe
acute congestion of the liver, slight ascites, free fibrin accumulations in the abdomen
(not the firm capsular adhesions seen in passive congestion), and distended portal
lymphatics.
Obstruction of the extrahepatic portal vein is quite uncommon. External compression
may occur because of adjacent abscesses or neoplasms. Portal vein thrombosis may be
caused by damage to the portal vein by local inflammatory processes or be associated
with states of hypercoagulability or retrograde intravascular growth of hepatic neoplasms.
In dogs, thrombosis of the portal vein has been associated with distant neoplasia,
immune-mediated hemolytic anemia, protein-losing nephropathy and enteropathy, pancreatitis,
peritonitis, and corticosteroid administration (Fig. 2-49
). Portal vein obstructions of slow development are expected to lead to portal hypertension
and its consequences. Atresia or hypoplasia of the extrahepatic portal vein can be
demonstrated in some cases of primary portal vein hypoplasia and is discussed previously
in the section on Developmental disorders.
Figure 2-49
Portal vein thrombosis in a dog.
Obstruction of intrahepatic portal vessels is a consequence of progressive fibrosing
lesions of primary hepatic disease centered on the portal triads. The small portal
vessels may be obliterated in the proliferative portal lesions, and new connections
may be established, including small functional arteriovenous communications. Regenerative
hepatic nodules and neoplasms may deform and compress portal vessels in some locations.
Portal hypertension is defined as increased blood pressure within the portal vein
caused by resistance to normal flow rates. Portal hypertension can arise from disturbances
of venous blood flow in any of the following 3 sites: prehepatic, intrahepatic, and
posthepatic.
Prehepatic portal hypertension is relatively uncommon and occurs when blood flow through
the portal vein is impaired before it enters the liver. The most common cause is portal
vein thrombosis. Intrahepatic portal hypertension arises from increased resistance
to blood flow at the level of the hepatic parenchyma. Intrahepatic portal hypertension
can be subdivided into 3 forms: presinusoidal (including intrahepatic portal vein
hypoplasia, intrahepatic arterioportal fistulae, periportal neoplastic or inflammatory
infiltrates, and periportal fibrosis), sinusoidal (cirrhosis or long-term inflammatory
disease with fibrosis and capillarization of sinusoids, amyloidosis, neoplastic infiltrations),
and postsinusoidal (perivenular fibrosis, veno-occlusive disease).
Posthepatic causes of portal hypertension are uncommon, but can result from increased
resistance to blood flow in the major hepatic veins, caudal vena cava, or right heart.
Causes include partial or complete thrombosis of the hepatic veins, which is very
rare; obstruction of the hepatic veins by neoplastic occlusion (e.g., pheochromocytoma);
and kinking of the caudal vena cava, associated with abdominal trauma. Congestive
heart failure can also lead to portal hypertension.
Regardless of cause, persistent portal hypertension can lead to acquired portosystemic
shunts (discussed later), with the exception of passive congestion, which rarely,
if ever, results in the development of shunt vessels. These shunts are usually numerous
and composed of distended thin-walled veins, which may connect the mesenteric veins
and the caudal vena cava. Ascites is common in conditions that develop acquired shunts
because of the associated portal hypertension.
Efferent hepatic vessels
Efferent flow begins in the sinusoids and passes through terminal hepatic venules
and larger hepatic veins to the vena cava. In the sinusoidal network, flow is interconnected
in many directions. However, in normal conditions, lobule outflow is closely matched
to inflow. In dogs, hepatic veins have substantial spiral smooth-muscle sphincters
that regulate outflow, but these are not evident in other domestic species. Splanchnic
engorgement during anesthesia and anaphylaxis is an indication that these smooth muscles
control canine hepatic outflow in a dynamic manner. Obstruction at the level of the
large hepatic veins can occasionally occur because of suppurative phlebitis, hepatic
abscesses, neoplasms, or other physical obstacles that impinge on the hepatic veins.
Similar space-occupying lesions in or around the vena cava in the diaphragm or mediastinum
can also affect hepatic outflow, along with systemic venous return.
The amount and arrangement of conventional connective tissue around the terminal hepatic
veins and larger hepatic veins influence the patterns of fibrosis observed in various
patterns of centrilobular injury. Hypoxia from anemia, heart failure, or shock can
cause centrilobular necrosis, as can many toxic agents that are preferentially injurious
to zone 3 hepatocytes. Necrosis at this level elicits a tissue repair response involving
the fibrous connective tissue along the terminal hepatic venules. Tissue fibrosis
can increase the resistance of the hepatic parenchyma and slow local outflow. This
can redirect sinusoidal blood to alternative less-affected venules, unless the necrosis
is extensive. Hepatic veno-occlusive disease
is the obliteration of small intrahepatic veins that may begin with damage to the
sinusoidal endothelium, accumulation of red cells and fibrin in the subintimal space,
and subsequent subendothelial fibrosis. This pattern of postnecrotic fibrosis can
contribute to the development of portal hypertension. Veno-occlusive disease is a
feature of pyrrolizidine toxicosis in humans ingesting these alkaloids in so-called
bush tea. In domestic animals, occlusive changes in terminal venules are notable in
poisoning by ragwort (Senecio jacobea) in cattle and also in dogs treated experimentally
with the pyrrolizidine alkaloid monocrotaline. Veno-occlusive disease has also been
reported as a consequence of prolonged chemotherapy, radiotherapy, and bone marrow
transplantation in humans, and similarly in dogs treated with irradiation or busulfan.
Idiopathic veno-occlusive disease with perivenular fibrosis around central and sublobular
veins, causing Budd-Chiari–like syndrome (see later), has also been reported as a
rare occurrence in the dog and cat. Total or subtotal obliteration of central hepatic
or sublobular veins by subintimal accumulation of collagen and fibrous tissue has
also been reported in captive snow leopards and cheetahs.
Passive congestion of the liver develops when the blood pressure in the hepatic veins
increases relative to that of the hepatic portal veins and can occur in any species.
It is almost always the consequence of cardiac dysfunction, including acute and chronic
heart failure, pericardial disease, and processes that obstruct the flow of blood
into or out of the heart, such as abscesses, heartworm disease, and local neoplasia.
Right-sided heart failure, in particular, produces elevated pressure within the caudal
vena cava that later involves the hepatic vein and its tributaries.
Acute passive congestion of the liver occurs when there is sudden cardiac decompensation,
particularly on the right side of the heart, or shock. Grossly, there is slight enlargement
of the liver, which is typically dark red, and blood flows freely from any cut surface
(Fig. 2-50
). The intrinsic lobular pattern of the liver may be slightly more pronounced, particularly
on the cut surface, because centrilobular areas are congested (dark red) in contrast
to the more normal color of the remainder of the lobule. The microscopic picture in
acute passive congestion is initially characterized by distention of central veins
and adjacent sinusoids, with accompanying distension of lymphatics in the stroma of
hepatic veins, portal triads, and the capsule. The appearance of the liver differs
with the duration and severity of the congestion. Fatty change, trabecular atrophy,
and necrosis of centrilobular hepatocytes with retention of the perisinusoidal reticulum
framework develop quickly. Erythrocytes tend to move into the perisinusoidal spaces
left by the lost hepatocytes, and may be seen trapped there when blood drains from
sinusoids and veins in freshly fixed sections. The intrahepatic network of lymphatics
at this stage becomes very distended and may form extensive cavernous channels about
hepatic veins and venules, in portal triads, and just beneath the capsule. With increased
venous pressure, transudation of red blood cells and high-protein-content edema can
develop, leading to polymerization of released fibrinogen on the capsular surface
that yields a fibrin coating of the capsule and blood-tinged abdominal fluid. Distension
of the space of Disse may be seen, but rarely, if ever, in tissue well fixed soon
after death, and is best regarded as a postmortem artifact. By comparison, perisinusoidal
edema is more obvious in hepatic congestion associated with shock or inflammatory
conditions.
Figure 2-50
Acute passive congestion of the liver in a cria with a congenital heart defect.
In chronic passive congestion, the amounts of fibrous connective tissue in the liver
increase in various patterns that differ among species. The capsular surface becomes
thicker and more opaque, and can develop a finely nodular texture with the formation
of capsular plaques (eFig. 2-11A). In dogs and cats, the edges of the central lobes
become rounded, whereas the margins of lateral and caudate lobes are sharpened by
peripheral atrophy and fibrosis. If the cause of the congestion is still present,
there is usually copious ascites at this stage. In species in which fibrosis is more
pronounced, a chronically congested liver has a distinct reticulated acinar pattern,
often more obvious beneath the capsule than in deeper parenchyma. This pattern is
known as “nutmeg liver” and is the result of the contrast of red centrilobular zones
of congestion with loss of hepatocytes, among pale swollen periportal parenchyma composed
of viable hepatocytes that are fatty (eFig. 2-11B). This classic pattern of chronic
passive congestion is most obvious in ruminants and horses. The “nutmeg” pattern can
be mimicked by some forms of toxic periacinar necrosis or fatty change but should
always be distinguishable from them by the presence of fibrous plaques in Glisson's
capsule in the passively congested liver. The pale lobular pattern is less obvious
in carnivores and should not be expected in animals that have insufficient adipose
reserves for mobilization. Over longer periods, centrilobular fibrosis links terminal
hepatic venules with one another and with the larger portal triads in a pattern known
as cardiac fibrosis.
eFigure 2-11
A. Chronic passive congestion in the liver of a cat. B. Cut surface of a bovine liver
with chronic passive congestion associated with traumatic reticulopericarditis.
(Courtesy K.G. Thompson.)
Inflammatory changes in the outflow veins are sometimes observed. Acute inflammation
and thrombosis of sublobular veins are typical of acute salmonellosis in many species,
but these changes are terminal and not associated with hepatic dysfunction. Inflammatory
cell infiltrates are occasionally observed in the larger hepatic veins, and have been
described in dogs with parvoviral myocarditis. Muscular hypertrophy of walls of hepatic
veins has been described in dogs with arteriovenous fistulae. Fibrous remodeling of
these veins has been associated with nitrosamine intoxication in domestic animals.
The term Budd
-
Chiari syndrome is used to describe the clinical features associated with hepatic
venous outflow obstruction caused by thrombosis of the main hepatic veins in humans.
Hepatic vein thrombosis is a rare occurrence in dogs and cats. In veterinary medicine,
it is more appropriate to use specific morphologic diagnostic terms to describe pathologic
changes that develop in response to various mechanical causes of postsinusoidal obstruction
of hepatic venous flow, resulting in development of hepatomegaly, postsinusoidal portal
hypertension, high-protein ascites, and acquired portosystemic collateral shunting.
Causes include obstruction of flow caused by tumors (e.g., intraluminal leiomyosarcoma
or other sarcoma, adrenal pheochromocytoma with extensive invasion into the caudal
vena cava in the dog) or abscesses in the liver or caudal vena cava, hepatic venous
or vena caval thrombosis, congenital malformation (fibrous web), kink or acquired
stricture occluding the lumen of the vena cava or hepatic veins (intrahepatic postsinusoidal
venous obstruction), or cardiac abnormalities impairing right atrial function (cor
triatrium dexter, neoplasia). Histologic changes include distension of the hepatic
veins and perivenous sinusoidal congestion, which, with chronicity, leads to perivenous
fibrosis typical of chronic passive congestion.
Thrombosis of the caudal vena cava has been described in detail in cattle, where rupture
of a hepatic abscess into the caudal vena cava is the most common etiology (Fig. 2-51
). Sequelae include pulmonary emboli, endoarteritis, multifocal pulmonary abscessation,
and chronic suppurative bronchopneumonia.
Figure 2-51
Thromboembolism of the caudal vena cava in an ox.
(Courtesy K.G. Thompson.)
Acquired portosystemic shunts
Congenital shunts have been described previously in the section on Developmental disorders.
Acquired portosystemic shunts within and external to the liver can develop in association
with various chronic liver diseases that lead to portal hypertension. These shunts
tend to be multiple, small, and tortuous (Fig. 2-52
). They may be difficult to identify postmortem when they are collapsed. They are
easily destroyed by routine dissection, so they must be noted before removal of the
abdominal viscera. Acquired shunts can be associated with evidence of portal hypertension,
such as distension of the portal veins and ascites, but when shunts are multiple and
well established, portal hypertension is somewhat relieved. It is important to distinguish
these varicose dilations that develop in response to portal hypertension from true
congenital portosystemic shunts. Acquired shunts arise in vestigial nonfunctional
portosystemic communications that dilate and become functional in response to portal
hypertension. They tend to develop between mesenteric veins and the caudal vena cava,
right renal vein, or gonadal veins, and are multiple, taking the form of a plexus
of tortuous, thin-walled vessels. Esophageal shunts and varicosities are common in
humans with cirrhosis, but are much less important in domestic animals. This may be
related to postural differences such that shunts are more likely to enter the vena
cava, where central venous pressure is least. In dogs, acquired shunts are most numerous
along the caudal mesentery.
Figure 2-52
Acquired shunt vessels in a dog.
Peliosis hepatis/telangiectasis
Peliosis hepatis is a term used to designate a hepatic vascular disorder characterized
by cystic, blood-filled spaces in the liver. The distinction between telangiectasis
and peliosis hepatis is subtle at best, and both conditions can be considered together.
There are 2 forms of peliosis hepatis, although both can appear in the same animal.
Intrinsic weakness of the local reticulin framework leading to sinusoidal dilation
is the hallmark of the phlebectatic form of peliosis hepatis, and the other form is
characterized by death of hepatocytes and sinusoidal distention (parenchymal type).
Peliosis hepatis lesions occur throughout the liver as dark red areas, irregular in
shape but well circumscribed, and ranging from pinpoints to many centimeters in size
(Fig. 2-53
, eFig 2-12). Sectioned or capsular surfaces are depressed after death, and on cutting,
they appear as cavities from which the blood drains to reveal a delicate network of
residual stroma. The histologic lesion is characterized by multiple, dilated, blood-filled
spaces sometimes surrounded by fibromyxoid stroma. The blood-filled cystic spaces
may be lined by endothelium. In humans, these lesions have an idiosyncratic association
with administration of various drugs, including anabolic and contraceptive steroids.
Infectious peliosis hepatis caused by Bartonella henselae or B. quintana infection
has also been described in immunosuppressed human patients.
Figure 2-53
Peliosis hepatis (telangiectasis) in the liver of an ox.
(Courtesy K.G. Thompson.)
eFigure 2-12
Peliosis hepatis in the liver of a cat.
(Courtesy K.G. Thompson.)
Peliosis hepatis has been reported in cattle, dogs, and cats, but the pathogenesis
is unknown. B. henselae DNA has been identified by PCR from the liver of a single
canine case, but does not seem to be involved in the feline disorder. Bovine hepatic
peliosis hepatis (telangiectasis) and human peliosis hepatis have been suggested to
share a similar pathogenesis. Primary alteration of the sinusoidal barrier, with rupture
of the reticulin fibers and increased deposition of basement membrane components in
the perisinusoidal region and fibrosis, may alter oxygen and substrate exchange between
hepatocytes and the blood, leading to hemodynamic imbalance, hepatocyte atrophy, and
eventually to sinusoidal disruption. Peliosis hepatis in livers is quite common in
older cats and cattle (Fig. 2-54
). There is no evidence clinically of related liver dysfunction. In cats, the cavities
are rather more frequent in the subcapsular zone and rarely exceed 2-3 mm in size.
There are often other senile changes in these livers, such as chronic fatty change,
nodular hyperplasia, and chronic hepatitis.
Figure 2-54
Histologic appearance of peliosis hepatis in a dog.
A specific condition termed “peliosis” develops in cattle poisoned by plants of Pimelea
spp. This form begins as diffuse periportal sinusoidal dilation. Because these changes
are also found in these animals in the spleen and in other organs with sinusoidal
microcirculation, it seems that the lesions may be adaptive to progressive and dramatic
increases in total blood volume. In the late stages of the intoxication by Pimelea,
the liver may resemble a huge, blood-filled sponge (Fig. 2-55
). The animals eventually die of a combination of hemodilutional anemia and circulatory
failure.
Figure 2-55
Periportal sinusoidal dilation resulting from chronic Pimelea poisoning in an ox.
(Courtesy R. Kelly.)
Further reading
Arey LB. Throttling veins in the livers of certain mammals. Anat Rec 1941;81:21-33.
Brown PJ, et al. Peliosis hepatis and telangiectasis in 18 cats. J Small Anim Pract
1994;35:73-77.
Buchmann AU, et al. Peliosis hepatis in cats is not associated with Bartonella henselae
infections. Vet Pathol 2010;47:163-166.
Cave TA, et al. Idiopathic hepatic veno-occlusive disease causing Budd-Chiari-like
syndrome in a cat. J Small Anim Pract 2002;43:411-415.
Crawford JM, Burt AD. Anatomy, pathophysiology and basic mechanisms of disease. In:
Burt AD, et al., editors. Macsween's Pathology of the Liver. 6th ed. New York: Churchill
Livingstone; 2012. p. 2-77.
Eipel C, et al. Regulation of hepatic blood flow: the hepatic arterial buffer response
revisited. World J Gastroenterol 2010;16:6046-6057.
Epstein RB, et al. A canine model for hepatic venoocclusive disease. Transplantation
1992;54:12-16.
Fine DM, et al. Surgical correction of late-onset Budd-Chiari-like syndrome in a dog.
J Am Vet Med Assoc 1998;212:835-837.
Gunn C, et al. Hepatic dearterialization in the dog. Am J Vet Res 1986;47:170-175.
Kalt DJ, Stump JE. Gross anatomy of the canine portal vein. Anat Histol Embryol 1993;22:191-197.
Kelly WR. The pathology and haematological changes in experimental Pimelea spp. poisoning
in cattle (“St. George disease”). Aust Vet J 1975;51:233-243.
Kelly WR, Seawright AA. Pimelea poisoning of cattle. In: Keeler RF, et al., editors.
Effects of Poisonous Plants on Livestock. New York: Academic Press; 1978. p. 293-300.
Kitchell BE, et al. Peliosis hepatis in a dog infected with Bartonella henselae. J
Am Vet Med Assoc 2000;216:519-523.
Marcato PS, et al. Pretelangiectasis and telangiectasis of the bovine liver: a morphological,
immunohistochemical and ultrastructural study. J Comp Pathol 1998;119:95-110.
Seawright AA. Phlebectatic peliosis hepatis in Australian cattle. Vet Hum Toxicol
1984;26:208-213.
Schoeman JP, Stidworthy MF. Budd-Chiari-like syndrome associated with an adrenal phaeochromocytoma
in a dog. J Small Anim Pract 2001;42:191-194.
Shulman HM, et al. Induction of hepatic veno-occlusive disease in dogs. Am J Pathol
1987;126:114-125.
Thompson JS, et al. Adequate diet prevents hepatic coma in dogs with Eck fistulas.
Surg Gynecol Obstet 1986;162:126-130.
Van Winkle TJ, Bruce E. Thrombosis of the portal vein in eleven dogs. Vet Pathol 1993;30:28-35.
Yamamoto K. Ultrastructural study on the venous sphincter in the sublobular vein of
the canine liver. Microvasc Res 1998;55:215-222.
Inflammatory Diseases of the Liver and Biliary Tract
The inflammatory response in the liver is unusual for 3 main reasons. First, hepatic
microvasculature is structurally and functionally different from tissues with capillary
vasculature. Microvascular permeability to plasma proteins, a hallmark of acute inflammation
in most tissues, is a normal property of the fenestrated sinusoidal endothelium of
the liver. Thus sinusoidal edema is not a prominent feature of acute parenchymal inflammation,
although edema can be observed in the capsule, portal tracts, connective tissue of
the terminal and sublobular hepatic veins (particularly in dogs), and in the wall
of the gallbladder. Microvascular blood flow in hepatic sinusoids is also less responsive
to the actions of various vasoactive mediators that alter blood flow in most other
acutely inflamed tissues. Second, resident Kupffer cells play an important and complex
role in liver inflammation, injury, and repair. Kupffer cells function in the innate
immune response, acting as the final component of the gut barrier by phagocytosing
pathogens, immunoreactive material, and endotoxin entering the liver via the portal
circulation. However, Kupffer cells also exhibit a range of different activated phenotypes,
depending on the local metabolic and immune environment. Classically activated macrophages
secrete proinflammatory cytokines, including tumor necrosis factor-α, interleukin-1
(IL-1), IL-6, IL-12, and inducible nitric oxide synthase (iNOS), influencing cell
populations in the liver and elsewhere, whereas alternatively, activated macrophages
express anti-inflammatory mediators, including IL-10 and contribute to resolution
of inflammation and promote repair. Dysregulation of the complex control of inflammatory
responses in Kupffer cells can contribute to chronic inflammation in the liver. Third,
the liver has central regulatory influences on many proinflammatory insults and inflammatory
mediators. As the major source of acute-phase proteins, including secreted pathogen
recognition receptors (PRRs), short pentraxins, components of the complement system,
and regulators of iron metabolism, hepatocytes are essential constituents of innate
immunity and largely contribute to the control of a systemic inflammatory response.
The liver is also the site of degradation of most soluble plasma proteins, and Kupffer
cells are the main site of clearance of immune complexes from the circulation, playing
an important role in the development of immunotolerance to potential antigenic substances
absorbed from the intestine. A full discussion of immune surveillance, and the complex
innate and adaptive immune responses involved in initiation and regulation of liver
inflammation, is beyond the scope of this chapter, and readers are directed to current
review articles on the subject.
These unusual aspects of the inflammatory response in the liver can make it more difficult
to differentiate certain degenerative and inflammatory conditions in this organ. Ongoing
cell death and repair can appear inflammatory, and acute leukocyte responses can cause
necrosis and apoptosis in the liver. The term
“necroinflammatory”
is convenient when the underlying pathogenetic mechanisms of necrosis and inflammation
are unknown.
Increased numbers of leukocytes (sinusoidal leukocytosis) are observed in hepatic
sinusoids in many acute or subacute bacteremias, as well as in conditions of steroid
excess in dogs. This change can be diagnostically useful but does not constitute evidence
of hepatitis unless there is obvious infiltration of granulocytes, monocytes, or lymphocytes
into the perisinusoidal space. Extramedullary hematopoiesis can also appear as focal
aggregates of myeloid cells in the perisinusoidal compartment. This change can be
distinguished from inflammatory infiltrates by the presence of immature myeloid cells.
Occasionally, hematopoietic cells from the splenic red pulp can be artifactually extruded
into the portal vasculature and appear in the liver, usually as nucleated cell aggregates
in the larger portal veins.
Infectious agents capable of causing hepatitis include viruses, bacteria, fungi, protozoa,
and helminths. Autoimmune and idiosyncratic drug responses also occur, but often the
etiology of acute or chronic hepatitis cannot be determined.
Hepatic inflammation
The liver is subject to infectious and degenerative insults that elicit inflammatory
responses in various patterns, for which the general term hepatitis is appropriate.
The term hepatitis
is used for focal or diffuse hepatic conditions that are either caused by infectious
agents or characterized by a leukocytic infiltrative inflammatory response, irrespective
of the cause. This definition allows inclusion of viral infections that are hepatotropic,
even though the lesions are mainly characterized by hepatocellular necrosis or apoptosis
rather than by the inflammatory response to the agent. The term hepatitis is also
used for responses to some hepatic toxicants, metals, or drug metabolic idiosyncrasies
in which there is a prominent leukocytic response to damaged cells. However, in responses
in which single necrotic hepatocytes elicit a mild neutrophilic or histiocytic response,
the term hepatitis is less appropriate because the pattern of injury is primarily
degenerative. Cholangitis
refers to inflammation of the biliary tree
. More specifically, choledochitis refers to inflammation of the bile ducts, and cholecystitis
refers to inflammation of the gallbladder. Cholangiohepatitis
applies to hepatic inflammation centered on the biliary tract and extending into adjacent
hepatic parenchyma.
Patterns and character of inflammation in hepatitis vary according to causative agent,
severity and stage of disease, the route of entry into the liver, and pathogenesis
of liver injury. Some viral pathogens, such as canine adenovirus 1, can cause acute
and diffuse hepatitis, with centrilobular to widespread hepatocellular necrosis, mixed
leukocyte infiltrates, sinusoidal congestion, and edema. By comparison, most infectious
causes of hepatitis, for example, toxoplasmosis, various herpesviruses, and various
bacteria, produce a more patchy pattern of inflammation with focally intense leukocyte
and Kupffer cell responses in the vicinity of areas of necrosis. The distribution,
character, and chronicity of these focal lesions are important diagnostically, but
they typically do not damage enough functional hepatic parenchyma, ducts, or vasculature
to produce systemic signs of liver failure. Foci of hepatitis with necrosis are common
incidental findings, and these are assumed to reflect localized responses to bacteria
that arrive via the portal system. Occasionally, such focal necroinflammatory lesions
are large and numerous, for example, in cattle with rumenitis.
Acute hepatitis
Hallmarks of acute hepatitis typically include a combination of inflammation, typically
a granulocytic inflammatory cell infiltrate, foci of hepatocellular apoptosis and
necrosis, and in some instances, evidence of regeneration. Leukocyte infiltrates in
acute diffuse hepatitis tend to accumulate mainly in the vicinity of the portal tracts,
around major bile ducts, or sometimes in the capsule and around the central veins.
Small but important numbers of neutrophils and mononuclear cells, including lymphocytes,
are usually seen in the perisinusoidal space and among hepatocytes, and are often
focally concentrated in sites of necrosis, or adjacent to infectious organisms. Edema
is an unusual feature of acute hepatitis, but is seen in severe injury, for example,
in infectious canine hepatitis. Grossly, hepatic edema is most obvious in the gallbladder,
large extrahepatic bile ducts, hepatic lymph nodes, and sometimes in the capsule.
Microscopically, edema is also evident in the portal triads, in the connective tissue
of the terminal and sublobular veins, and sometimes by an increase in the perisinusoidal
space.
Kupffer cells are key participants in the acute inflammatory responses in the liver.
They can enlarge and accumulate vacuoles and lysosomal debris during regular phagocytic
removal of microorganisms, cell debris, and extravascular erythrocytes. They can also
be activated to secretory histiocytes that release various cytokines and other mediators
that induce hypertrophic or proliferative responses of hepatocytes, stellate cells,
and endothelium. Activated Kupffer cells are larger and more prominent or numerous
in sections, their nuclei are larger and vesicular, and their cytoplasm is basophilic
and may contain vacuoles or ingested particulate matter. In overwhelming infections,
many of the Kupffer cells and adjacent sinusoidal endothelial cells and hepatocytes
undergo necrosis.
Chronic hepatitis
Chronic liver disease in domestic animals has historically been classified into several
different entities based on morphologic criteria, but fibrosis is a consistent feature.
In human medicine, classification of chronic hepatitis has been simplified, and morphologic
divisions such as chronic active hepatitis and chronic persistent hepatitis, originally
defined in specific clinical contexts, have been abandoned because of problems of
evolving definitions and application, and lack of correspondence with prognosis. The
single designation “chronic hepatitis” is now used for chronic necroinflammatory disease
lasting more than 6 months, further modified by specifying the etiology, type and
severity of inflammation, and degree and distribution of apoptosis and/or necrosis
(disease activity or grade), and the degree of fibrosis (disease chronicity or stage).
Numerous systems for grading and staging chronic hepatitis have been developed. In
human medicine, chronic hepatitis is usually the result of chronic infection with
hepatotropic viruses, and, less commonly, autoimmune, drug-induced, or associated
with inherited metabolic diseases, such as Wilson disease. In veterinary medicine,
chronic liver disease may develop following chronic bile duct obstruction, infection
with hepatotropic infectious agents, familial or hereditary metabolic diseases, or
may be toxic, drug-induced, or possibly autoimmune in origin. However, the majority
of chronic liver disease is idiopathic, reflecting deficiencies in our current level
of understanding of the etiologic, pathophysiologic, and clinical implications of
the patterns of inflammation and necrosis seen in domestic animals.
Regardless of the etiology, initial acute liver damage will not progress to fibrosis
or cirrhosis unless the inflammation and damage are protracted, for example, by ongoing
hepatocellular injury mediated by immunologic mechanisms, including antibody- and
lymphocyte-mediated cytotoxicity, or ongoing oxidative damage. Clinical signs are
nonspecific in the early stages, but as the disease progresses to involve more of
the liver and impair regeneration, icterus, ascites, and hepatic encephalopathy may
develop as typical correlates with hepatic insufficiency.
Chronic hepatitis of humans has several characteristic lesions. These are also evident
in animals, although not always to the same degree or frequency as described in the
various forms of viral, immune-mediated, and idiosyncratic hepatitis in humans. The
first is periportal interface hepatitis, sometimes referred to as “piecemeal necrosis”
(Fig. 2-56
). This necroinflammatory change initially destroys the limiting plate of periportal
hepatocytes, and may continue to erode into the hepatic parenchyma, expanding the
portal areas. Portal inflammation is variable in intensity, and includes infiltration
by lymphocytes and plasma cells. Bridging necrosis, with tracts of necrosis dissecting
across the hepatic lobule between portal triads or between portal areas and central
veins, may also develop. Degenerative changes affecting hepatocytes in areas of interface
hepatitis include cell swelling and apoptosis. Bile duct degeneration, multifocal
necrosis, and hepatocellular regeneration, in the form of 2-cell–thick hepatic plates
and mitotic figures, may also be seen. Deposition of collagen and basement membrane
material in the space of Disse leads to capillarization of hepatic sinusoids. Single
or small groups of hepatocytes may be isolated and entrapped in expanded portal areas.
Fibrosis may progress to bridge portal tracts and central veins, disrupting hepatic
lobular architecture and culminating in the development of cirrhosis.
Figure 2-56
Periportal interface hepatitis in chronic hepatitis in a dog.
Further reading
Batts KP, Ludwig J. Chronic hepatitis: an update on terminology and reporting. Am
J Surg Pathol 1995;19:1409-1417.
Beighs V, Trautwein C. The innate immune response during liver inflammation and metabolic
disease. Trends Immunol 2013;34:446-452.
Bode JG, et al. Hepatic acute phase proteins—regulation by IL-6 and IL-1 cytokines
involving STAT3 and its crosstalk with NF-kB-dependent signaling. Eur J Cell Biol
2012;91:496-505.
Dixon LJ, et al. Kupffer cells in the liver. Compr Physiol 2013;3:785-797.
Goodman ZG. Grading and staging systems for inflammation and fibrosis in chronic liver
diseases. J Hepatol 2007;47:598-607.
Ishak K, et al. Histological grading and staging of chronic hepatitis. J Hepatol 1995;22:696-699.
Ishak KG. Pathologic features of chronic hepatitis. Am J Clin Pathol 2000;113:40-55.
Knodel RG, et al. Formulation and application of a numerical scoring system for assessing
histological activity in asymptomatic chronic active hepatitis. Hepatol 1981;1:431-435.
Knolle PC, Thimme R. Hepatic immune regulation and its involvement in viral hepatitis
infection. Gastroenterol 2014;146:1193-1207.
Ludwig J. The nomenclature of chronic active hepatitis: an obituary. Gastroenterol
1993;105:274-278.
Sterczer A, et al. Chronic hepatitis in the dog—a review. Vet Q 2001;23:148-152.
Szabo G, Csak T. Inflammasomes in liver diseases. J Hepatol 2012;57:642-654.
Chronic hepatitis in dogs
Chronic hepatitis is a relatively common diagnosis in dogs, characterized by hepatocellular
apoptosis or necrosis, a variable mononuclear or mixed inflammatory cell infiltrate,
evidence of regenerative attempts, and fibrosis of variable extent and pattern. Although
various infectious etiologies and drug- and toxin-induced chronic hepatic disease
have been described, the majority remains idiopathic, although excess hepatic copper
accumulation is the best characterized cause of chronic hepatitis in the dog. Breeds
of dogs reported to be at increased risk for chronic hepatitis include the Bedlington
Terrier, Doberman Pinscher, West Highland White Terrier, Labrador Retriever, American
and English Cocker Spaniel, Skye Terrier, Standard Poodle, Dalmatian, and English
Springer Spaniel. Hepatic copper accumulation in association with chronic hepatitis
has been documented in most of these breeds, but apart from the recognized genetic
mutation in the COMMD1 gene in Bedlington Terriers, leading to a primary copper storage
disease with impaired copper excretion, the pathogenesis of the breed-related copper
accumulation remains unclear.
In the dog, disease grade is correlated with increasing hepatocellular apoptosis,
proliferation, expression of nitric oxide synthase isoforms, and total hepatic iron,
whereas disease stage is correlated with increasing α-smooth muscle actin–labeled
periductular myofibroblasts and perisinusoidal stellate cells, CK7-positive ductular
cells, the extracellular matrix protein tenascin-C, and expression of genes important
in the production and regulation of hepatic fibrosis, including platelet-derived growth
factors PDGFB and PDGFD, thrombospondin 1, transforming growth factors β1 and β2 (TGFB1
and TGFB2), matrix metalloproteinase 2 and tissue inhibitor of matrix metalloproteinase
1, and the collagen genes COL1A1 and COL3A1.
Given the importance of autoimmune hepatitis in human medicine, several studies have
evaluated the possible contributions of immune mechanisms in dogs. Upregulation of
major histocompatibility complex class II antigen expression in hepatocytes has been
demonstrated in Doberman hepatitis in association with lymphocyte infiltration, and
it has been proposed that hepatocytes presenting a putative major histocompatibility
complex class II molecule–associated autoantigen could be targets for T-cell–mediated
immune attack. An association with specific major histocompatibility complex DLA class
II haplotypes has been described in Doberman Pinschers and English Springer Spaniels
with subclinical or clinical hepatitis, and it has been suggested that the highly
polymorphic DLA genes may be involved in altered susceptibility to chronic hepatitis.
However, a primary autoimmune pathogenesis has yet to be demonstrated.
The presence, mechanism, and role of hepatic copper accumulation in dogs continues
to generate ongoing interest. Although copper is an essential metal cofactor for cuproenzymes,
free copper ions can catalyze the formation of reactive hydroxyl radicals capable
of causing oxidative injury (see the Copper section later in Toxic hepatic disease).
The liver is central to copper homeostasis, and biliary excretion is the major route
for regulating levels of copper in the body. Hepatic copper accumulation can arise
(1) as the result of a primary metabolic defect in hepatic copper metabolism, (2)
secondary to abnormal hepatic function with cholestasis and altered biliary copper
excretion, or (3) as a consequence of excess dietary copper intake. Secondary copper
accumulation occurs in the majority of human patients with primary biliary cirrhosis,
prolonged extrahepatic bile duct obstruction, or chronic liver disease; however, dogs
appear to be relatively resistant to hepatocellular copper accumulation as a result
of cholestasis, at least in experimental studies following ligation of the common
bile duct.
In the dog, the reference range for hepatic copper is generally considered to be ≤400 µg/g
dry weight (DW);.concentrations >1,800-2,000 µg/g DW are considered pathogenic. In
primary genetic copper storage disorders and excess copper intake, copper accumulation
appears at least initially to be primarily centrilobular, extending throughout the
lobule as the condition progresses, with hepatic copper concentrations usually >2,000 µg/g
DW. In copper storage secondary to abnormal hepatic function, copper usually accumulates
in smaller amounts in the periportal parenchyma or without a consistent pattern, in
concentrations typically <2,000 µg/g DW.
When the hepatic concentration of copper surpasses 400 µg/g DW, the excess begins
to accumulate within lysosomes. These copper-laden lysosomes become consistently demonstrable
by the histochemical stains rubeanic acid and rhodanine, at copper concentrations
exceeding 400 µg/g DW (Fig. 2-57
), although some studies note visible copper granules at hepatic copper concentrations
as low as 200 µg/g DW. A reasonable estimate of copper burden can be made from stained
sections; however, biochemical determination of copper is more reliable and can be
useful to follow the effectiveness of copper chelation therapy. Care should be taken
when collecting liver for copper levels, as once the normal lobular architecture is
lost because of injury and regenerative nodule formation, copper distribution within
the liver becomes heterogeneous. Hepatocytes in regenerative nodules often have relatively
low levels of copper, possibly as an adaptation that facilitates their ability to
proliferate and form nodules, or as a function of inadequate time for significant
copper accumulation in the hepatocytes forming nodules. The intervening areas of parenchymal
collapse may contain an abundance of copper. Because of this regional variation, small
samples, such as those taken from needle biopsies, are often inaccurate, and larger
samples are required.
Figure 2-57
Copper staining (rhodanine) in the liver of a West Highland White Terrier.
Regardless of the reasons for copper accumulation, lysosomal copper can exceed a threshold
or be released when hepatocytes die, and thereby contribute to the development of
hepatitis. Hepatic pathology does not typically occur at concentrations <2,000 µg/g
DW, although higher values can still be found in some dogs with normal liver histology,
and other factors, such as antioxidant levels and other oxidative stress from drugs
or other factors, may influence the relationship between copper concentration and
injury.
A hereditary, autosomal recessive copper-associated hepatopathy associated with impaired
biliary copper excretion and progressive accumulation of copper within hepatocytes
has been well documented in Bedlington Terriers.
The majority of affected Bedlington Terriers have been found deficient in a protein,
COMMD1 (copper metabolism MURR1 domain protein 1), caused by deletion of exon 2 in
the COMMD1 gene, although there are other less common mutations that can also occur.
Although its function is not fully understood, the COMMD1 protein has been shown to
interact with the copper transporter ATP7B, important in Wilson disease, a copper
storage disorder in humans, and its absence in affected dogs may impair ATP7B-mediated
copper export from hepatocytes into the canaliculus. Homozygous affected dogs have
the highest copper levels. Bedlington Terriers are the only breed to date shown to
accumulate copper continuously throughout life, and hepatic copper concentrations
in these animals may be very high; as much as 12,000 µg/g DW has been recorded, and
levels >5,000 µg/g are common. Affected dogs are usually presented with signs of progressive
liver failure, including ill-thrift, wasting, ascites, and signs of encephalopathy.
An acute form may occur in some dogs, with acute hepatic necrosis and release of copper
into the systemic circulation, where it provokes a hemolytic crisis and rapidly developing
anemia and icterus (eFig. 2-13). The initial histologic lesion is multifocal centrilobular
hepatitis, with foci of macrophages, lymphocytes, plasma cells, and neutrophils among
the copper-laden hepatocytes in zone 3. Apoptotic hepatocytes, some containing copper
granules, appear at the periphery of some foci. Copper levels >3,000 µg/g DW result
in widespread massive necrosis in some dogs. Survivors may progress to develop postnecrotic
cirrhosis. Grossly, the livers in later stages are fibrotic, pale, and finely nodular.
eFigure 2-13
Icteric Bedlington Terrier with an acute hemolytic crisis concurrent with chronic
copper-associated hepatitis.
(Courtesy University of Guelph.)
Chronic hepatitis is well documented in Doberman Pinschers.
The disease is more common in middle-aged female dogs. Histologic changes in the livers
of dogs exhibiting clinical signs of advanced hepatic disease include piecemeal necrosis
of periportal zone 1 hepatocytes, with a mixed inflammatory cell infiltrate, as well
as necrosis of zone 3 hepatocytes with bridging necrosis crossing the lobule (Fig.
2-58
). Copper accumulation is evident in centrilobular hepatocytes, with various degrees
of portal fibrosis, bile duct proliferation, bridging fibrosis, and development of
cirrhosis in the most severely affected dogs. Intrahepatocyte bile pigment accumulation
and intracanalicular bile stasis are present in periportal zones, along with iron
accumulation in Kupffer cells and macrophages.
Figure 2-58
Periportal interface hepatitis with mixed inflammatory infiltrates and bridging portal
fibrosis in a Doberman Pinscher dog.
Elevated concentrations of hepatic copper have been reported in many, but not all,
Doberman Pinschers with chronic hepatitis, and the significance of increased hepatic
copper concentration in this breed remains controversial. A recent investigation into
the possibility of impaired copper excretion in affected Doberman Pinschers reported
that 5 dogs with elevated liver copper and persistent subclinical hepatitis but without
demonstrable cholestasis had comparable rates of plasma copper clearance to control
dogs, but reduced rates of biliary excretion of 64Cu, suggesting that impaired copper
excretion may play a role in disease in this breed. Significantly reduced levels of
mRNA of various proteins involved in copper binding, transport, and excretion, including
ATP7A, ATP7B, ceruloplasmin, and metallothionein, have been documented in Dobermans
with clinical hepatitis with high hepatic copper concentrations, along with a reduction
in gene expression of components of the antioxidant defense system, including SOD1,
catalase, as well as reduced levels of glutathione, so continued investigations are
needed to further clarify the pathogenesis of chronic hepatitis in Dobermans.
West Highland White Terriers are at increased risk of developing chronic hepatitis
and cirrhosis. There is evidence to support familial hepatic copper accumulation in
some West Highland White Terriers, although the mode of inheritance is not completely
understood. Hepatic copper accumulates in zone 3 hepatocytes, up to ~8 months of age,
with concentrations rarely >2,000 µg/g DW. Clinical illness directly attributable
to copper hepatotoxicity (concentrations >2,000 µg/g DW) in West Highland White Terriers
is, however, apparently uncommon. Idiopathic chronic hepatitis progressing to cirrhosis
does also occur in this breed, and may be distinguished on the basis of a different
zonal location and morphology of the inflammatory lesions. In the idiopathic disease,
inflammatory foci are smaller, composed of a single apoptotic hepatocyte or fragments
of cells accompanied by a few lymphocytes and plasma cells, and are commonly localized
to zone 1, or may be random in distribution. In dogs with copper toxicosis, foci of
inflammation and necrosis were larger, always found around the central vein among
copper-laden hepatocytes, and were composed of debris-filled macrophages, lymphocytes,
plasma cells, and scattered neutrophils, with occasional apoptotic hepatocytes around
the periphery. Distinguishing between copper toxicosis and idiopathic chronic hepatic
disease may be difficult in cirrhotic livers, which, irrespective of the underlying
cause, may have reduced copper burdens because of connective-tissue displacement of
hepatic parenchyma, and typically lower concentrations of copper in regenerative nodules.
Chronic hepatitis been reported in genetically related Skye Terriers,
accompanied by modest and somewhat inconsistent hepatic copper accumulation. Lesions
ranged from hepatocellular degeneration and necrosis with mild inflammation in zone
3, to chronic hepatitis and cirrhosis with marked intracanalicular cholestasis. Hepatic
copper concentrations ranged from 800-2,200 µg/g DW, and copper-containing hepatocytes
were found predominantly in zone 3.
Copper-associated chronic hepatitis has been described in Labrador Retrievers,
characterized by centrilobular infiltrates of macrophages containing intracytoplasmic
copper and hemosiderin, fewer neutrophils, mononuclear inflammatory cells, as well
as scattered foci of hepatocellular necrosis, lobular collapse, periacinar to bridging
fibrosis, nodular regeneration, and in some cases cirrhosis. Hepatic copper concentrations
in one study were typically >2,000 µg/g, whereas mean hepatic copper concentrations
in a separate study were also found to be significantly higher in affected dogs (614 µg/g,
range 104-4,234 µg/g) compared to age- and sex-matched control dogs (299 µg/g, 93-3,810 µg/g).
A concurrent general increase in hepatic copper concentrations in a study population
of Labrador Retrievers spanning 30 years was postulated to be associated with increased
dietary copper availability, the result of a change in the form and bioavailability
of supplemental copper added to commercially produced dog foods. A positive association
between dietary copper levels and hepatic copper concentrations has been demonstrated,
and high dietary copper has been suggested as a risk factor for development of copper-associated
hepatitis in susceptible animals within the breed. Concurrent renal proximal tubular
dysfunction with glucosuria, and increased renal copper has been described in some
Labrador Retrievers with copper-associated hepatitis, and sporadically in other breeds.
Chronic liver disease associated with elevated hepatic copper concentrations has also
been reported in Dalmatians.
A range of necroinflammatory changes has been reported, including multifocal, piecemeal,
centrilobular to massive hepatic necrosis, and cirrhosis, although, in one study of
10 dogs, various degrees of piecemeal necrosis and bridging fibrosis were the most
common histologic change, with either primarily lymphocytic or neutrophilic inflammatory
infiltrates. Morphologic or biochemical evidence of cholestatic liver disease was
not prominent. Hepatic copper concentrations ranged from 745-8,390 µg/g DW (mean 3,197 µg/g)
in one report of 10 dogs, aged 2-10 years, with a variable zonal distribution. Three
previous cases in young Dalmatians reported hepatic copper concentrations of 7,940 µg/g
DW, 1,916 µg/g wet weight, and 2,356 µg/g wet weight, and 2 of these reports describe
diffuse positive staining for copper in all hepatocytes, with the strongest staining
observed in centrilobular hepatocytes.
Chronic hepatitis has been reported in American and English Cocker Spaniels and English
Springer Spaniels,
characterized clinically in the later stages by ascites, weight loss, and icterus.
Affected dogs develop chronic hepatitis and cirrhosis, and, at postmortem, livers
are typically small and firm, with multiple small regenerative nodules (Fig. 2-59
). Histologically, there is moderate to severe portal hepatitis, with inflammatory
infiltrates of predominantly lymphocytes, plasma cells, and fewer neutrophils, and
variable degrees of portal fibrosis and bridging fibrosis. One study of American Cocker
Spaniels reported diffuse fibrosis typical of lobular dissecting hepatitis in 7 of
13 affected dogs, in addition to patterns of fibrosis more typical of cirrhosis. Piecemeal
necrosis and limiting plate destruction have been reported in Cocker Spaniels, whereas
in English Springer Spaniels, hepatocyte necrosis and apoptosis in areas of inflammatory
infiltrates in both portal areas and scattered throughout the hepatic parenchyma was
more typical. Biliary hyperplasia or marked ductular reaction was noted in affected
Cocker Spaniels. Hepatic copper staining is variable, and hepatic copper accumulation
is not a consistent feature.
Figure 2-59
Chronic hepatitis in an American Cocker Spaniel.
α1-Antitrypsin (α1-AT) deficiency
has been suggested to play a role in chronic hepatitis in some dog breeds, including
English Cocker Spaniels, although whether this is an epiphenomenon or cause of chronic
liver disease has not yet been proven. α1-AT, a plasma glycoprotein synthesized mainly
by hepatocytes, is a member of the serine proteinase inhibitor (serpin) superfamily,
and is a potent inhibitor of neutrophil elastase. Hereditary α1-AT deficiency in humans
is associated with mutations that perturb the protein's tertiary structure and promote
polymerization. These misfolded forms of α1-AT accumulate within the rough endoplasmic
reticulum of hepatocytes, forming PAS-positive globules. α1-AT deficiency has been
associated in humans with neonatal hepatitis, juvenile cirrhosis, and adult hepatocellular
carcinoma.
Lobular dissecting hepatitis, associated with predominantly sinusoidal inflammation
and fibrosis, has been described in young dogs with ascites and acquired portosystemic
shunts. The liver is usually small, pale with a predominantly smooth surface, and
occasional hyperplastic nodules (eFig. 2-14). The histologic lesion is characterized
by dissection of lobular parenchyma by reticulin and fine collagen fibers into individual
and small groups of hepatocytes, accompanied by a variable, mixed inflammatory infiltrate
of lymphocytes, plasma cells, and lesser numbers of neutrophils and macrophages (Fig.
2-60A, B
). Activated fibroblastic cells, likely hepatic stellate cells, may also be prominent
along sinusoids. Hepatocytes form rosettes or pseudoductular structures, and regenerative
nodules may be present. Portal inflammation and periportal fibrosis are not conspicuous
features of this disease. The etiology of this disorder remains unknown.
Figure 2-60
A. Lobular dissecting hepatitis with diffuse fine interstitial fibrosis isolating
hepatocytes in a juvenile Golden Retriever dog. B. Reticulin stain demonstrating pattern
of lobular dissection.
eFigure 2-14
Lobular dissecting hepatitis in a dog.
(Courtesy J.L. Caswell.)
Chronic hepatitis in other species
Copper-associated liver disease has also been described in domestic cats. A hepatopathy
with excess hepatic copper (4,074 µg/g DW) was first reported in a Siamese cat, characterized
by enlarged, finely vacuolated, and individual necrotic hepatocytes with centrilobular
and midzonal copper accumulation. Chronic hepatitis and cirrhosis, with marked accumulation
of stainable copper in macrophages in fibrous septa and with sparser staining in regenerative
nodules, was reported in a European Shorthair cat with similarly elevated liver copper
concentrations (4,170 µg/g DW; the upper reference range limit for hepatic copper
concentration is cats is <180 µg/g DW). A recent retrospective study described an
additional 11 cats with presumed primary copper-associated hepatopathy, characterized
by elevated hepatic copper concentrations (>700 µg/g DW), diffuse or centrilobular
and intermediate zone copper staining, without any other co-occurring cholestatic
disorders. Affected cats had hepatocyte vacuolation consistent with glycogen accumulation,
fibrillar collagen in the perivenular region, and variable mild parenchymal collapse,
with bridging fibrosis in one cat. Cats also accumulate copper in the liver secondary
to other cholestatic hepatobiliary disorders, such as chronic cholangitis/cholangiohepatitis,
or extrahepatic bile duct obstruction, although copper staining in these cases is
principally in hepatocytes in portal and intermediate zones.
Chronic hepatitis seen as end-stage livers occurs in horses (eFig. 2-15) and ruminants,
particularly cattle, and is often assumed to be associated with ingestion of hepatotoxins,
such as pyrrolizidine alkaloids in forage, or a consequence of prolonged administration
of hepatotoxic therapeutics, although in the absence of specific histologic lesions
and/or a corroborative clinical history, the underlying causes are rarely discovered.
eFigure 2-15
Chronic hepatitis with cirrhosis in a horse.
Further reading
Andersson M, Sevelius E. Breed, sex and age distribution in dogs with chronic liver
disease: a demographic study. J Small Anim Pract 1991;32:1-5.
Azumi N. Copper and liver injury: experimental studies on the dogs with biliary obstruction
and copper loading. Hokkaido Igaku Zasshi 1982;57:331-349.
Bexfield NH, et al. Chronic hepatitis in the English springer spaniel: clinical presentation,
histological description and outcome. Vet Rec 2011;169:415-419.
Bexfield NH, et al. DLA class II alleles and haplotypes are associated with risk for
and protection from chronic hepatitis in the English Springer Spaniel. PLoS ONE 2012;7:e42854.
Boisclair J, et al. Characterization of the inflammatory infiltrate in canine chronic
hepatitis. Vet Pathol 2001;38:628-635.
Boomkens SY, et al. Hepatitis with special reference to dogs. A review on the pathogenesis
and infectious etiologies, including unpublished results of recent own studies. Vet
Q 2004;26:107-114.
Center SA. Chronic liver disease: current concepts of disease mechanisms. J Small
Anim Pract 1999;40:106-114.
Center SA, et al. Digital image analysis of rhodanine-stained liver biopsy specimens
for calculation of hepatic copper concentrations in dogs. Am J Vet Res 2013;74:1474-1480.
Dill-Macky E. Chronic hepatitis in dogs. Vet Clin N Am Small Anim Pract 1995;25:387-398.
Dyggve H, et al. Association of Doberman hepatitis to canine major histocompatibility
complex II. Tissue Antigens 2010;77:30-35.
Favier RP, et al. Copper-induced hepatitis: the COMMD1deficient dog as a translation
animal model for human chronic hepatitis. Vet Q 2011;31:49-60.
Fieten H, et al. Association of dietary copper and zinc levels with hepatic copper
and zinc concentration in Labrador Retrievers. J Vet Intern Med 2012;26:1274-1280.
Fieten H, et al. Canine models of copper toxicosis for understanding mammalian copper
metabolism. Mamm Genome 2012;23:62-75.
Fuentealba C, Aburto EM. Animal models of copper-associated liver disease. Comp Hepatol
2003;2:5-16.
Fuentealba C, et al. Chronic hepatitis: a retrospective study in 34 dogs. Can Vet
J 1997;38:365-373.
Haynes JS, Wade PR. Hepatopathy associated with excessive hepatic copper in a Siamese
cat. Vet Pathol 1995;32:427-429.
Haywood S, et al. Pathobiology of copper-induced injury in Bedlington terriers: ultrastructural
and microanalytical studies. Anal Cell Pathol 1996;10:229-241.
Hoffman G. Copper-associated liver diseases. Vet Clin Small Anim 2009;39:489-511.
Hoffmann G, et al. Copper-associated chronic hepatitis in Labrador Retrievers. J Vet
Intern Med 2006;20:856-861.
Hurwitz BM, et al. Presumed primary and secondary hepatic copper accumulation in cats.
J Am Vet Med Assoc 2014;244:68-77.
Jensen AL, Nielson OL. Chronic hepatitis in three young standard poodles. J Vet Med
A 1991;38:194-197.
Johnston AN, et al. Hepatic copper concentrations in Labrador Retrievers with and
without chronic hepatitis: 72 cases (1980-2010). J Am Vet Med Assoc 2013;242:372-380.
Kanemoto H, et al. Expression of fibrosis-related genes in canine chronic hepatitis.
Vet Pathol 2011;48:839-845.
Kanemoto H, et al. American Cocker Spaniel chronic hepatitis in Japan. J Vet Intern
Med 2013;27:1041-1048.
Klomp AEM, et al. The ubiquitously expressed MURR1 protein is absent in canine copper
toxicosis. J Hepatol 2003;39:703.
Langlois DK, et al. Acquired proximal renal tubular dysfunction in 9 Labrador Retrievers
with copper-associated hepatitis (2006-2012). J Vet Intern Med 2013;27:491-499.
Mandigers PJ, et al. Hepatic 64Cu excretion in Dobermanns with subclinical hepatitis.
Res Vet Sci 2007;83:204-209.
Mandigers PJ, et al. Chronic hepatitis in Doberman pinschers. A review. Vet Q 2004;26:98-106.
Mandigers PJ, et al. Association between liver copper concentration and subclinical
hepatitis in Doberman Pinschers. J Vet Intern Med 2004;18:647-650.
Meertens NM, et al. Copper-associated chronic hepatitis and cirrhosis in a European
Shorthair cat. Vet Pathol 2005;42:97-100.
Mekonnen GA, et al. Tenascin-C in chronic canine hepatitis: immunohistochemical localization
and correlation with necro-inflammatory activity, fibrotic stage, and expression of
alpha-smooth muscle actin, cytokeratin 7, and CD3+ cells. Vet Pathol 2007;44:803-813.
Noaker LJ, et al. Copper associated acute hepatic failure in a dog. J Am Vet Med Assoc
1999;214:1502-1505.
Poitout F, et al. Cell-mediated immune responses to liver membrane protein in canine
chronic hepatitis. Vet Immunol Immunopathol 1997;57:169-178.
Poldervaart JH, et al. Primary hepatitis in dogs: a retrospective review (2002-2006).
J Vet Intern Med 2009;23:72-80.
Rolfe DS, Twedt DC. Copper-associated hepatopathies in dogs. Vet Clin North Am Small
Anim Pract 1995;25:399-417.
Rutgers HC, et al. Idiopathic hepatic fibrosis in 15 dogs. Vet Rec 1993;133:115-118.
Sevelius E, et al. Hepatic accumulation of α1-antitrypsin in chronic liver disease
in the dog. J Comp Pathol 1994;111:401-412.
Smedley R, et al. Copper-associated hepatitis in Labrador Retrievers. Vet Pathol 2009;46:484-490.
Spee B, et al. Differential expression of copper-associated and oxidative stress related
proteins in a new variant of copper toxicosis in Doberman pinschers. Comp Hepatol
2005;4:3-14.
Spee B, et al. Copper metabolism and oxidative stress in chronic inflammatory and
cholestatic liver diseases in dogs. J Vet Intern Med 2006;20:1085-1092.
Speeti M, et al. Lesions of subclinical Doberman hepatitis. Vet Pathol 1998;35:361-369.
Speeti M, et al. Upregulation of major histocompatibility complex class II antigens
in hepatocytes in Doberman hepatitis. Vet Immunol Immunopathol 2003;96:1-12.
Sterczer A, et al. Chronic hepatitis in the dog—a review. Vet Q 2001;23:148-152.
Stockhaus C, et al. A multistep approach in the cytologic evaluation of liver biopsy
samples of dogs with hepatic diseases. Vet Pathol 2004;41:461-470.
Thornburg LP. Histomorphological and immunohistochemical studies of chronic active
hepatitis in Doberman Pinschers. Vet Pathol 1998;35:380-385.
Thornburg LP. A perspective on copper and liver disease in the dog. J Vet Diagn Invest
2000;12:101-110.
Thornburg LP, et al. Hepatic copper concentrations in purebred and mixed-breed dogs.
Vet Pathol 1990;27:81-88.
Thornburg LP, et al. The relationship between hepatic copper content and morphologic
changes in the liver of West Highland white terriers. Vet Pathol 1996;33:656-661.
Twedt DC, et al. Clinical, morphological and chemical studies on copper toxicosis
of Bedlington terriers. J Am Vet Med Assoc 1979;175:269-275.
van De Sluis BJ, et al. Genetic mapping of the copper toxicosis locus in Bedlington
terriers to dog chromosome 10, in a region syntenic to human chromosome region 2p13-p16.
Hum Mol Genet 1999;8:501-507.
van De Sluis B, et al. Identification of a new copper metabolism gene by positional
cloning in a purebred dog population. Hum Mol Genet 2002;15:165-173.
van den Ingh TSGAM, et al. Morphological classification of parenchymal disorders of
the canine and feline liver. In: Rothuizen J, et al., editors. WSAVA Standards for
Clinical and Histological Diagnosis of Canine and Feline Liver Diseases. Chapt. 7.
Philadelphia: Saunders Elsevier; 2006. p. 85-101.
van den Ingh TSGAM, Rothuizen J. Lobular dissecting hepatitis in juvenile and young
adult dogs. J Vet Intern Med 1994;8:217-220.
Vince AR, et al. Hepatic injury correlates with apoptosis, regeneration, and nitric
oxide synthase expression in canine chronic liver disease. Vet Pathol 2013;51:932-945.
Webb CB, et al. Copper-associated liver disease in Dalmatians: a review of 10 dogs
(1998-2001). J Vet Intern Med 2002;16:665-668.
Whittemore JC, et al. Hepatic copper and iron accumulation and histologic findings
in 104 feline liver biopsies. J Vet Diagn Invest 2012;24:656-661.
Miscellaneous inflammatory liver disease
Nonspecific reactive hepatitis refers to hepatic inflammation characterized by light
inflammatory infiltrates, primarily in the portal tracts, without any evidence of
hepatocellular necrosis. It is often observed during inflammation of the splanchnic
organs and is a reactive response, rather than primary inflammation of the liver.
Giant cell hepatitis
is an uncommon lesion in animals, but is recorded in cats, calves, and foals (Fig.
2-61
). There is evidence for maternal leptospirosis in some cases reported in foals. Two
cases are reported in young cats with concurrent thymic lymphomas. The liver is deeply
bile stained. Histologically, the acinar structure is effaced, and the blood vessels
engorged. Hepatocytes are large and syncytial and may contain 10 or more nuclei. The
pale or ballooned cytoplasm contains bile pigments, and cytoplasmic invaginations
into hepatocyte nuclei are common. Inflammatory cells are not conspicuous. Liver parenchymal
giant cell transformation occurs in humans in a wide variety of congenital and neonatal
liver disorders, including bile duct obstruction associated with biliary atresia,
viral and bacterial infections, some metabolic disorders such as galactosemia, some
cases of Down syndrome and other genetic disorders, as well as in idiopathic neonatal
hepatitis, a cholestatic condition of undetermined cause. Although giant cells were
originally suggested to be a marker of infantile obstructive cholangiopathy in humans,
their association with a wide range of disorders supports an alternative conclusion
that giant cell formation represents a nonspecific reaction of the infant's hepatocytes
to various types of injury.
Figure 2-61
Giant cell hepatitis in a cat.
Hypertrophic hepatic cirrhosis in calves is described from Germany and occasionally
observed elsewhere. Death may occur in liver failure within a few days to weeks of
birth, or the disease may be discovered at slaughter for veal. The liver is moderately
enlarged with rounded borders, very firm but smooth on the surface, and gray. Histologically,
there is some biliary hyperplasia, but the lesion is dominated by diffuse fibrosis
infiltrated in the early stages by mononuclear inflammatory cells. No cause has been
identified. Similar lesions of congenital hepatic fibrosis may be found in unborn
or aborted calves.
Systemic granulomatous disease has been reported in cattle grazing hairy vetch (Vicia
villosa). The disease is characterized clinically by dermatitis, pruritus, diarrhea,
wasting, and high mortality. Histologic lesions include infiltration of skin and internal
organs, including portal areas of the liver, by monocytes, lymphocytes, plasma cells,
eosinophils, and multinucleated giant cells. The pathogenesis is unknown, although
the inflammatory reaction has characteristics of a type IV hypersensitivity reaction.
Alternatively, vetch lectin may act to activate T lymphocytes directly to initiate
the cellular response.
Further reading
Messer NT, Johnson PJ. Idiopathic acute hepatic disease in horses: 12 cases (1982-1992).
J Am Vet Med Assoc 1994;204:1934-1937.
Panciera RJ, et al. Hairy vetch (Vicia villosa Roth) poisoning in cattle: update and
experimental induction of the disease. J Vet Diagn Invest 1992;4:318-325.
Poonacha KB, et al. Leptospirosis in equine fetuses, stillborn foals, and placentas.
Vet Pathol 1993;30:362-369.
Suzuki K, et al. Giant cell hepatitis in two cats. J Vet Med Sci 2001;63:199-201.
Inflammatory diseases of the biliary tract
Inflammation of the gallbladder is termed
cholecystitis.
Inflammation of the large bile ducts is
cholangitis,
whereas inflammation of the smallest intrahepatic bile ductules is termed
cholangiolitis. Cholangiolitis is uncommon in animals, and occurs mainly in conjunction
with inflammation of larger ducts. Destructive cholangiolitis has been observed in
dogs, and is associated with adverse drug reactions in humans.
Cholecystitis
Cholecystitis is uncommon and is often associated with concurrent cholelithiasis,
although acalculous cholecystitis has been reported in the dog, with a single case
report in a pig. Cholecystitis is thought to be caused by reflux of intestinal bacteria
into the gallbladder via the bile ducts, or to hematogenous entry of bacteria from
the adjacent hepatic circulation. Aerobic gram-negative bacteria are the most frequent
isolates from canine cases, although occasionally anaerobic bacteria such as clostridia
have been cultured. Campylobacter jejuni has been isolated from 2 dogs with bacteremia
and cholecystitis. Occasionally, parasites, such as flukes that colonize the bile
ducts, can enter the gallbladder and cause cholecystitis. Canine cholecystitis has
been associated with various systemic disorders, including diabetes mellitus, severe
enteritis, biliary stasis, septicemia, as well as with the use of immunosuppressive
drugs, each of which may promote bacterial colonization of the gallbladder. In the
acute lesion, histologic changes include neutrophilic inflammatory infiltrates in
the wall and lumen of the gallbladder, with focal erosion or ulceration, and edema.
More chronic stages develop typical mixed inflammatory infiltrates, with fibrosis.
Occasionally, the infiltrate may be predominantly lymphoplasmacytic, with formation
of lymphoid follicles within the mucosa.
Gallbladder infarction characterized by transmural coagulative necrosis of the gallbladder
wall with intravascular fibrin thrombi has been reported in dogs (Fig. 2-62
). The arterial blood supply to the gallbladder is the cystic artery, a branch of
the hepatic artery, and occlusion may cause partial or complete infarction. Predisposing
factors have not been identified; however, the lack of significant concurrent inflammatory
response suggests that this is not simply a sequela to underlying cholecystitis.
Figure 2-62
Infarcted gallbladder in a dog.
Gallbladder mucocele is discussed further under Ectopic, hyperplastic, and metaplastic
lesions.
Cholangitis/cholangiohepatitis
Although pure cholangitis does occur, extension of inflammation from the ducts into
the adjacent hepatic parenchyma can occur, and such lesions can quite accurately be
regarded as cholangiohepatitis.
Bile contains various antimicrobial factors, including β-defensins and bile acids
that are inhospitable to most bacteria, except for some species with particular capsule
adaptations. Bacterial cholangiohepatitis is usually caused by common opportunists
of enteric origin, such as coliforms and streptococci. Some bacteremic organisms,
including some Salmonella, can be cultured from the bile, but the mechanism by which
they get there is unknown. Salmonellosis is a distinctive cause of fibrinous cholecystitis
in cattle, especially calves.
The pathogenesis of bacterial cholangitis/cholangiohepatitis depends on various predisposing
conditions, similar to those involved in the pathogenesis of pyelonephritis. These
include infection by bacteria that reach the ducts hematogenously, facilitated by
localization in the peribiliary plexus or by extension from sinusoidal Kupffer cells
or foci of necrosis. Descending cholangitis is occasionally observed in cattle with
suppurative hepatitis with extension from abscesses directly into the ducts or via
the portal lymphatics. Cholangiohepatitis of this origin may be restricted in its
distribution to biliary fields, but in some cases, it does become quite diffuse in
the biliary system. Alternatively, bacteria can ascend the ducts from the intestine,
facilitated by bile stasis caused by mechanical or functional obstructions. The inflammatory
and proliferative responses in cholangitis further interfere with bile flow and exacerbate
the ductular spread of bacterial infections once they are established. Again, these
lesions can be quite regionally variable, and may be missed on hepatic biopsy if only
small areas are sampled.
The course and pathologic changes in cholangiohepatitis vary greatly, from fulminating
suppurative infection to persistent mild inflammation that, over a period of months
or years, leads to hepatic fibrosis of biliary distribution. Severe suppurative cholangiohepatitis
may follow a short course to death, the effects being those of the infection itself,
which may become septicemic, rather than of hepatic injury. At autopsy, the liver
is swollen, soft, and pale, and its architecture is blurred. Few or many suppurative
foci may be visible beneath the capsule and on the cut surface (eFig. 2-16). They
are small, sometimes miliary in distribution, and not encapsulated. Lesions in other
organs may be those of septicemia and jaundice. Microscopically, the larger ducts
contain purulent exudate, and the smaller ones are disintegrated. Dense masses of
neutrophils, liquefied or not, are present in the portal triads and infiltrate the
degenerate parenchyma.
eFigure 2-16
Cholangiohepatitis in a cat.
(Courtesy J.L. Caswell.)
In subacute and chronic cholangiohepatitis, the inflammation is more proliferative
than exudative. The liver is enlarged and may be of normal shape, or distorted because
of irregular areas of atrophy and regenerative hyperplasia. Its surface may be smooth
or finely granular; the capsule is thickened and may bear fibrous tags or be adherent
to adjacent viscera. Within areas of duct obstruction, retention of bile pigments
can be found in the regions served by the occluded ducts, but systemic icterus (or
photosensitization in herbivores) is unlikely unless a large portion of liver is affected.
On cut surface, the enlarged portal tracts are easily visible and accentuate the architecture
of the organ. Eventually, the new fibrous tissue replaces the parenchyma, and in chronic
diffuse cases in which the original infection persists, continuous fibroplasia may
produce hepatic enlargement, the organ becoming huge, gray, and gristly. This spectrum
of liver changes is typical of alsike clover poisoning (“big-liver disease”) in horses
(discussed in the later section on Toxic hepatic disease). Alternatively, the chronic
fibrosis may occur in wedge-shaped areas oriented to a small bile duct. The enlarged
interlobular ducts may be readily visible and frequently contain plugs of inspissated
secretion and debris.
Microscopically, the reaction remains centered on portal tracts. These are expanded
in subacute cases by infiltration of leukocytes and macrophages and the proliferation
of small ducts and, in chronic cases, chiefly by organizing fibrous tissue and proliferating
bile ducts. Encroachment on the parenchyma is minimal but inevitable. Continued degeneration
of the periportal parenchyma is probably an additional stimulus to local fibroplasia,
which, as well as thickening the smallest portal triads, extends along their length
and links up with neighboring triads, thus subdividing the acini into segments. The
hepatic venules and sublobular veins are involved. Regenerative nodules are not a
prominent feature of cholangiohepatitis unless large areas of parenchyma have been
destroyed, in which case the least damaged lobes are expanded by coarse nodules.
Neutrophilic cholangitis (suppurative or exudative cholangitis/cholangiohepatitis)
is a relatively common disorder of cats and, much less commonly, dogs. This condition
has been suggested to be the result of acute inflammation in the biliary tree associated
with ascending bacterial infection, and in cats, the condition often occurs in conjunction
with other disorders, including acute extrahepatic bile duct obstruction, pancreatitis,
or inflammatory bowel disease. In the cat, the biliary and pancreatic ducts share
a common entry to the duodenum, and simultaneous infectious inflammation of these
systems is common. Escherichia coli is the most frequent bacterial isolate; however,
Bacteroides, Klebsiella, hemolytic Streptococcus, and clostridia have also been reported.
The acute stage is characterized by edema and neutrophilic portal infiltrates, inflammation
and degeneration of bile ducts with neutrophils in ductular lumens and emigrating
through the ductular epithelium. Some cases may progress to cholangiohepatitis, with
infiltration of inflammatory cells into hepatic lobules and periportal hepatocellular
necrosis. Areas of suppuration and hepatic abscessation may occur. Mixed cholangiohepatitis,
with neutrophils, lymphocytes, and plasma cells infiltrating portal areas accompanied
by bile duct proliferation, biliary epithelial degeneration, and various degrees of
periportal to bridging fibrosis, may represent the subacute stage (Fig. 2-63
), whereas concentric periportal fibrosis and pseudolobule formation may represent
the most chronic stage of the disease.
Figure 2-63
Neutrophilic bile duct inflammation with portal edema, early fibrosis, and a mixed
infiltrate of neutrophils, lymphocytes and plasma cells extending across the limiting
plate in a case of subacute cholangiohepatitis in a dog.
An apparently distinct condition of cats,
lymphocytic cholangitis (lymphocytic portal hepatitis, nonsuppurative cholangitis/cholangiohepatitis)
is a slowly progressive disease characterized by lymphocytic infiltrates within the
portal area, with variable degrees of bile duct or oval cell proliferation and peribiliary,
portal-to-bridging fibrosis (Fig. 2-64
). An immune-mediated pathogenesis has been proposed, and in one study, most cats
with lymphocytic cholangitis had T-cell–predominant portal infiltrates, often with
accompanying portal B-cell aggregates. The presence of various degrees of lymphocytic
targeting and infiltration of bile ductules with concurrent degenerative changes in
ductular epithelium, destructive cholangitis leading to ductopenia, and portal lipogranulomas
may be useful in distinguishing this condition from hepatic lymphoma in the cat, along
with T-cell receptor clonality assays.
Figure 2-64
Lymphocytic cholangitis in a cat.
Destructive cholangitis has also been described in dogs, and is characterized by destruction
and loss of bile ducts in the smaller portal tracts, with subsequent inflammation
composed of pigment-laden macrophages, neutrophils, and/or eosinophils, and in some
instances, fibrosis. The extent of injury may be sufficient to cause marked icterus
and acholic feces because of marked intrahepatic cholestasis. The pathogenesis of
this lesion is unclear; however, toxic injury, idiosyncratic drug reactions (see Fig.
2-40) and infection by canine distemper virus have been implicated in some cases.
Biliary tract obstruction
Cholelithiasis
(gallstone formation) is seldom observed in animals. The choleliths usually form in
the gallbladder and are composed of a mixture of cholesterols, bile pigments, salts
of bile acids, calcium salts, and a proteinaceous matrix. Choleliths of mixed composition
are yellow-black or green-black and are friable. Pigment stones, composed of calcium
bilirubinate, and cholesterol stones have also been reported in dogs. There may be
hundreds of small stones or a few large ones. The large stones are usually faceted.
The origin of these mixed gallstones is uncertain, but their development is probably
secondary to chronic mild cholecystitis and related to disturbances of the resorptive
activities of the gallbladder, whereby the bile salts are removed faster than the
stone-forming compounds. Gallstones are usually asymptomatic. Occasionally, they lodge
in and obstruct bile ducts and cause jaundice. The larger stones may cause pressure
necrosis and ulceration of the mucosa, local dilations of the bile ducts, and saccular
diverticula of the gallbladder. Calculi seldom form in the ducts, although calcareous
deposits often do so in fascioliasis of cattle. Calcium bilirubinate calculi have
been reported in the bile ducts of horses (Fig. 2-65
), associated with intermittent jaundice.
Figure 2-65
Cholelith obstructing the bile duct in a horse.
Occasionally, particles of solid ingesta may find their way into the gallbladder;
sand has been seen in sheep, and seeds in pigs.
Biliary obstruction is rarely caused by impacted gallstones. Usually, it is the result
of cholangitis or cholecystitis, the obstruction being produced by masses of detritus
and biliary constituents, parasites, or cicatricial stenosis of the ducts. Adult ascarids
may cause mechanical obstruction. Inspissated bile-stained friable plugs are occasionally
responsible for obstructions in segments of the liver in horses. Tumors of the pancreas
and duodenum, and tumors and abscesses of the hilus of the liver and portal nodes,
may cause compression stenosis of the ducts. Edematous swelling of the papilla in
enteritis may also be of significance. Biliary obstruction by abnormal intraluminal
mucoid secretion (gallbladder mucocele) has been reported in dogs (see later section
on Ectopic, metaplastic, and hyperplastic lesions).
The consequences of biliary obstruction depend on the site and duration of the obstruction.
When the main duct is involved, there is jaundice. When one of the hepatic ducts is
involved, there is no jaundice, and depending on the efficiency of biliary collaterals,
there may be no pigmentation of the obstructed segments of liver. Increases in serum
γ-glutamyltranspeptidase and alkaline phosphatase usually occur when a sufficiently
large amount of the duct system is affected. The ducts undergo progressive cylindrical
dilation, which may be extreme. The smallest interlobular ducts and the cholangioles
proliferate. There is inflammation in the walls of the ducts and the portal triads,
and this is probably due in part to chemical irritation by bile acids but is largely
caused by secondary bacterial infections. These infections may be acute and purulent,
or low grade; in these cases, bacteria may not be easily cultured. The cholangiohepatitis
that almost inevitably follows has been described previously.
Rupture of the biliary tract or the gallbladder causes steady leakage of bile into
the peritoneal cavity, the omentum being unable to seal even small defects. The bile
salts are very irritating and may cause acute chemical peritonitis. The peritoneal
effusion that follows may remain sterile; more often it is infected by enteric bacteria,
and severe diffuse peritonitis ensues. This may be rapidly fatal, particularly if
clostridia are involved. Many perforations of the biliary tract are traumatic in origin;
however, in dogs, cholecystitis, gallbladder necrosis/infarction, and gallbladder
mucocele have all been associated with gallbladder rupture.
Further reading
Callahan Clark JE, et al. Feline cholangitis: a necropsy study of 44 cats (1986-2008).
J Feline Med Surg 2011;13:570-576.
Center SA. Diseases of the gallbladder and biliary tree. Vet Clin North Am Small Anim
Pract 2009;39:543-598.
Crews LJ, et al. Clinical, ultrasonographic, and laboratory findings associated with
gallbladder disease and rupture in dogs: 45 cases (1997-2007). J Am Vet Med Assoc
2009;234:359-366.
Day MJ. Immunohistochemical characterization of the lesions of feline progressive
lymphocytic cholangitis/cholangiohepatitis. J Comp Pathol 1998;119:135-147.
Gabriel A, et al. Suspected drug-induced destructive cholangitis in a young dog. J
Small Anim Pract 2006;47:344-348.
Gagne JM, et al. Histopathologic evaluation of feline inflammatory liver disease.
Vet Pathol 1996;33:521-526.
Holt DE, et al. Canine gallbladder infarction: 12 cases (1993-2003). Vet Pathol 2004;41:416-418.
Kirpensteijn J, et al. Cholelithiasis in dogs: 29 cases (1980-1990). J Am Vet Med
Assoc 1993;202:1137-1142.
Mayhew PD, et al. Pathogenesis and outcome of extrahepatic biliary obstruction in
cats. J Small Anim Pract 2002;43:247-253.
Newell SM, et al. Gallbladder mucocele causing biliary obstruction in two dogs: ultrasonographic,
scintigraphic and pathological findings. J Am Anim Hosp Assoc 1995;31:467-472.
O'Neill EJ, et al. Bacterial cholangitis/cholangiohepatitis with or without concurrent
cholecystitis in four dogs. J Small Anim Pract 2006;47:325-335.
Osumi T, et al. A case of recovery from canine destructive cholangitis in a miniature
dachshund. J Vet Med Sci 2011;73:937-939.
Oswald GP, et al. Campylobacter jejuni bacteremia and acute cholecystitis in two dogs.
J Am Anim Hosp Assoc 1994;30:165-169.
Ryu SH, et al. Cholelithiasis associated with recurrent colic in a Thoroughbred mare.
J Vet Sci 2004;5:79-82.
Starost MF, Burkholder TH. Acalculous and clostridial cholecystitis in a pig. J Vet
Diagn Invest 2008;20:527-530.
van den Ingh TSGAM, et al. Destructive cholangitis in seven dogs. Vet Q 1988;10:240-245.
van den Ingh TSGAM, et al. Morphological classification of biliary disorders of the
canine and feline liver. In: Rothuizen J, et al., editors. WSAVA Standards for Clinical
and Histological Diagnosis of Canine and Feline Liver Diseases. Philadelphia: Saunders
Elsevier; 2006. p. 61-76.
Warren A, et al. Histopathologic features, immunophenotyping, clonality and eubacterial
fluorescence in situ hybridization in cats with lymphocytic cholangitis/cholangiohepatitis.
Vet Pathol 2011;48:627-641.
Weiss DJ, et al. Relationship between inflammatory hepatic disease and inflammatory
bowel disease, pancreatitis, and nephritis in cats. J Am Vet Med Assoc 1996;209:1114-1116.
Weiss DJ, et al. Inflammatory liver disease. Semin Vet Med Surg (Small Anim) 1997;12:22-27.
Infectious Diseases of the Liver
Viral infections
Various systemic viral diseases may affect the liver. Adenoviral infections of lambs,
calves, and goat kids can cause multifocal hepatic necrosis and cholangitis, in addition
to pneumonia. Lymphohistiocytic hepatitis, with jaundice and various degrees of apoptosis
of hepatocytes, disorganization of hepatic plates, and perilobular stromal condensation,
is seen in some cases of porcine circovirus 2–associated disease (Fig. 2-66A, B
). Canid herpesvirus 1 infection in puppies and, more rarely, in adult dogs causes
disseminated focal necrosis and hemorrhages in parenchymal organs, including the liver,
with formation of amphophilic intranuclear inclusion bodies in epithelial cells of
kidney, lung, and liver. Similar microfoci of hepatic necrosis sometimes occur in
aborted or newborn foals with congenital equid herpesvirus 1 infections (Fig. 2-67
), in neonatal calves infected with bovine herpesvirus 1, in piglets with suid herpesvirus
1 infections, and in feline fetuses following intravenous feline herpesvirus 1 inoculation
of the pregnant queen. Feline coronavirus infection can cause granulomatous hepatitis
in some infected cats, as part of feline infectious peritonitis (Fig. 2-68
). Multifocal hepatic necrosis has been described in large felids and domestic cats
infected with highly pathogenic influenza A virus H5N1. Hepatocellular and Kupffer
cell necrosis with mild mononuclear portal inflammation was reported in outbreaks
caused by virulent systemic strains of feline calicivirus in cats in the United States
and United Kingdom. Systemic cowpox virus infection with foci of hepatic necrosis
containing immunoreactive cowpox viral antigen has been reported in a cat.
Figure 2-66
A. Hepatitis caused by porcine circovirus 2 (PCV-2) infection in a pig. B. Immunohistochemical
stain for PCV-2 antigen.
(Courtesy J. DeLay.)
Figure 2-67
Focal hepatic necrosis with intranuclear inclusion bodies and syncytial cells in congenital
equid herpesvirus 1 infection in a foal.
Figure 2-68
Pyogranulomatous hepatitis caused by feline infectious peritonitis virus infection.
The viral diseases discussed in more detail later are those in which the liver is
the major target organ, causing substantial hepatic injury that sometimes culminates
in hepatic failure. Unlike humans, in which several pathogenic hepatitis viruses from
various families are well described, few viruses that specifically target the liver
have been identified in the major domestic mammals. Hepadnaviruses have been found
in members of the squirrel family, including woodchucks and various species of ground
squirrels and arctic ground squirrels, as well as in birds, including some species
of ducks, geese, and herons. Infection with hepatitis E virus (HEV) genotypes 3 and
4 is common in domestic and wild pigs, and although clinical disease is not apparent
in infected swine, they may serve as reservoirs for this potentially zoonotic pathogen.
Lesions of mild multifocal lymphoplasmacytic hepatitis with focal hepatocellular necrosis
have been reported in pigs experimentally inoculated with HEV, but lesions in natural
infections are not recognized. Flaviviruses, including a nonprimate hepacivirus and
a GB-virus–like virus (pegivirus) have recently been described in horses, although
their potential clinical significance is unknown. A second novel pegivirus species
of horses, Theiler's disease–associated virus, is described in more detail later.
Infectious canine hepatitis
Canine adenovirus 1 (CAdV-1) infection is the cause of infectious canine hepatitis,
a severe liver disease in dogs; other canids, including coyotes, foxes, wolves; and
in bears. Vaccination has made the disease rare in many countries in which it was
endemic. Deaths from infectious canine hepatitis are usually sporadic, although small
outbreaks can occur among young dogs in kennels. Fatalities seldom occur among dogs
>2 years of age. In areas where the disease is not controlled by vaccination, it is
probable that most dogs in the general population contact CAdV-1 in the first 2 years
of life and suffer either inapparent infection or mild febrile illness with pharyngitis
and tonsillitis.
In more severe cases, there is vomiting, melena, high fever, and abdominal pain. There
may be petechiae on the gums; the mucous membranes are pale and occasionally slightly
jaundiced. Nonspecific nervous signs occur in a few cases. There is also a peracute
form of the disease in which the animal is found dead without signs of illness, or
after an illness of only a few hours. In convalescence, there may be a unilateral
or bilateral opacity of the cornea caused by corneal edema (so-called blue eye, a
type III hypersensitivity reaction with immune-complex–mediated corneal injury) (Fig.
2-69
), which disappears spontaneously.
Figure 2-69
Infectious canine hepatitis. Corneal edema or “blue-eye.”
(Courtesy North Carolina State University CVM.)
CAdV-1 has special tropism for endothelium, mesothelium, and hepatic parenchyma, and
it is injury to these that is responsible for the pathologic features of edema, serosal
hemorrhage, and hepatic necrosis. The histologic specificity of the lesions depends
on the demonstration of large, solid intranuclear inclusion bodies in endothelium
or hepatic parenchyma (Fig. 2-70
). Inclusions are occasionally observed in other differentiated cells but always have
the same morphologic and tinctorial features, being deeply acidophilic with a blue
tint.
Figure 2-70
Infectious canine hepatitis (canine adenovirus 1 infection). Intranuclear inclusion
bodies in numerous hepatocytes.
(Courtesy University of Guelph.)
The morbid picture of spontaneously fatal cases is usually distinct enough to allow
a diagnosis to be made at gross postmortem. Superficial lymph nodes are edematous,
slightly congested, and often hemorrhagic. Blotchy or paintbrush hemorrhages may be
present on intestinal and gastric serosae, and there is usually a small quantity of
clear or blood-stained fluid in the abdomen. Jaundice, if present, is slight. The
liver is slightly enlarged, with sharp edges, and is turgid and friable, sometimes
congested, with a fine, uniform, yellow mottling (eFig. 2-17A). Red strands of fibrin
can be found on its capsule, especially between the lobes. In the majority of cases,
the wall of the gallbladder is edematous (eFig. 2-17B), and may have intramural hemorrhages;
when edema is mild, it may be detected only in the attachments of the gallbladder.
eFigure 2-17
Infectious canine hepatitis. A. Mottled, congested liver. B. Edema and hemorrhage
of the gallbladder.
(Courtesy University of Guelph.)
Gross lesions in other organs are inconstant. Small hemorrhagic infarcts may be found
in the renal cortices of young puppies. Hemorrhages may occur in the lungs, and occasionally,
there are irregular areas of hemorrhagic consolidation in the caudal lobes. Hemorrhages
in the brain occur in a small percentage of cases, typically only grossly visible
in the midbrain and brainstem. Hemorrhagic necrosis of medullary and endosteal elements
occurs in the metaphyses of long bones in young dogs, and the hemorrhages are readily
visible through the thin cortex of the distal ends of the ribs.
At low magnification, the histologic changes in the liver are of centrilobular (periacinar)
zonal necrosis, resembling the zonal pattern of some acute hepatotoxicities. The susceptibility
of centrilobular parenchyma to necrosis in this disease is as yet unexplained. Close
to the portal triads, the hepatocytes may be nearly normal in appearance, except for
loss of basophilia and the presence of a scattering of inclusion bodies. In spontaneously
fatal cases, most of the parenchyma of the peripheral and central portions of the
lobules (acini) is dead, the hepatocytes having undergone coagulative necrosis, and
in some of these, ghosts of inclusion bodies may be detectable. The margin between
necrotic parenchyma and viable tissue is usually quite sharp, although in the viable
tissue, there are many individual hepatocytes undergoing apoptosis, most of them without
inclusion bodies. Fatty change is common. The dead cells do not remain long, so the
sinusoids become dilated and filled with blood. The reticulin framework remains intact,
an observation in keeping with the fact that, in recovered cases, restitution of the
liver is complete. Massive necrosis with collapse does not occur. As is typical of
severe centrilobular necrosis, the necrotic zones, initially eccentric areas about
hepatic venules, extend and link up to isolate portal units. Intranuclear inclusions
can be found in Kupffer cells in variable numbers. Many of the Kupffer cells are dead,
others are proliferating, and others are actively phagocytic in the removal of debris.
Leukocytic reactions in the liver are mild and are directed against the necrotic tissue;
mononuclear cells are present, but neutrophils, many degenerating, predominate. There
is some collection of bile pigment, but it is moderate, in keeping with the short
course of the disease.
Microscopic lesions in other organs are largely the result of injury to endothelium.
Inclusion bodies in endothelial cells can be difficult to find and are looked for,
with most profit in renal glomeruli, where endothelium is concentrated. Occasionally,
they are found in the epithelium of collecting tubules. When areas of hemorrhagic
consolidation of the lungs are present, there is hemorrhage, edema, and fibrin formation
in the alveoli, and in these consolidated areas, inclusions are often common in alveolar
capillaries and even in dying cells of the bronchial epithelium. Changes in the brain
are secondary to vascular injury. Hemorrhages, if present, are from capillaries and
small venules, and inclusions in endothelial nuclei can usually be found in vessels
that have bled. Other endothelial and adventitial cells are hyperplastic and mixed
with a few lymphocytes. Small foci of softening or demyelination may be present in
relation to the hemorrhages. Lymphoreticular tissues are congested, and inclusions
may be found in reticulum cells of follicles, in the red pulp of the spleen, and in
macrophages anywhere.
Following natural oronasal exposure, viral multiplication occurs in the tonsils and
leads to tonsillitis, which may be quite severe with extensive edema of the throat
and larynx. Fever accompanies the tonsillitis and precedes the viremic phase, which
lasts 4-8 days, accompanied by leukopenia. Hepatic necrosis develops at about day
7 of experimental infection; however, an immune response with adequate neutralizing
antibodies may clear the virus from the blood and limit the extent of hepatic damage.
In surviving animals, hepatic regeneration occurs rapidly, and there do not appear
to be any significant residual lesions. Small foci of hepatocellular necrosis may
still be present at 2 weeks, and foci of proliferated Kupffer cells may be detectable
for another week or 2. Progressive hepatic injury does not seem to follow the acute
phase of the natural disease, although chronic hepatitis has been experimentally reproduced
in partially immunized dogs challenged with the virus. Adenoviral antigen has been
detected immunohistochemically in occasional Kupffer cells in 5 dogs with a range
of chronic hepatic inflammatory lesions and in one dog with a patent ductus venosus;
however, a retrospective study of formalin-fixed paraffin-embedded liver from 45 dogs
with chronic hepatitis or cirrhosis failed to reveal CAdV-1 by either PCR or immunohistochemistry,
although the possibility that the virus initiates hepatic damage by provoking self-perpetuating
hepatitis could not be excluded. Focal interstitial nephritis occurs commonly, and
the cellular infiltrates are persistent but not functionally significant. They consist
of interstitial lymphocytic accumulations, especially about the corticomedullary junction
and in the stroma of the pelvis.
Corneal edema is a late development, corresponding temporally to increasing neutralizing
antibody titer. It may occur as early as day 7 of infection, but is usually delayed
to between 14 and 21 days. Viral antigen can be detected in these eyes by fluorescent
techniques, but not in the corneal structures. Inflammatory edema is present in the
iris, ciliary apparatus, and corneal propria, and inflammatory cells are abundant
in the filtration angle and iris. The infiltrates are principally plasma cells, and
there is evidence that the ocular lesion is a hypersensitivity reaction to circulating
immune complex deposition, with complement fixation, inflammatory cell chemotaxis,
and corneal endothelial damage resulting in an anterior uveitis with corneal stromal
edema.
Originally, it was assumed that the widespread tendency to hemorrhage in this disease
was because of leakage from damaged vascular endothelium, coupled with an inability
on the part of the damaged liver to replace clotting factors. Although these effects
play a role, it is now known that the exhaustion of clotting factors is in large part
the result of their accelerated consumption, as the widespread endothelial damage
is a potent initiator of the clotting cascade.
Wesselsbron disease
This disease is caused by Wesselsbron virus, an arthropod-borne flavivirus that, according
to serologic surveys, is widespread in Africa in various species of animals and birds.
Various Aedes mosquitoes are the vectors. Humans are also susceptible to clinical
and inapparent infection. The virus produces outbreaks of abortion and perinatal death
in sheep. Susceptible adults rarely show clinical signs but may have a biphasic febrile
response to infection; other clinical signs, when present, are of hepatitis and jaundice.
The lesions in lambs dying within 12 hours of birth consist mainly of widespread petechiae
and gastrointestinal hemorrhage; longer survival allows jaundice to develop, and the
liver becomes orange-yellow, enlarged, friable, and patchily congested. The bile in
the gallbladder becomes thick and dark in some cases, but this may be due more to
hemorrhage into the gallbladder than to hemolysis. Lymph nodes are rather constantly
enlarged, congested, and edematous.
The most characteristic histologic changes are seen in the liver. There are randomly
scattered foci of necrosis, with apoptosis and proliferation of sinusoidal lining
cells (Fig. 2-71
). Mononuclear cells and pigment-filled macrophages accumulate in the portal stroma
as well as in the sinusoids. In a variable proportion of cases, hepatocyte nuclei
may contain eosinophilic, irregular inclusions. These are not accompanied by as much
margination of nuclear chromatin as that associated with conventional viral inclusions,
and their significance is obscure. Wesselsbron viral antigen can be demonstrated in
necrotic acidophilic and degenerating hepatocytes and rarely in the inclusions. In
jaundiced animals, there may be considerable canalicular cholestasis; whether or not
this is the result of hemolysis does not appear to have been determined. Hepatocellular
proliferation is apparent in the less acute cases. Lymphoid follicles in lymph nodes
and spleen show pronounced lymphocyte necrosis and stimulation of lymphoblasts.
Figure 2-71
Focal necrosis and apoptosis in Wesselsbron disease in a lamb.
(Courtesy S. Youssef.)
Rift Valley fever
This is an arthropod-borne viral infection of ruminants and humans, in many respects
similar to Wesselsbron disease. Rift Valley fever virus (RVFV) is, however, responsible
for greater losses. Morbidity and mortality may occur in adult sheep; death sometimes
occurs in adult cattle, but it is chiefly a disease of the young, causing heavy mortality
among lambs, kids, and calves, and abortion in ewes, does, and cows. The infection
in enzootic form is widespread in eastern and southern Africa, but it has, in plague-like
proportions, extended to West Africa, Egypt, and the Arabian Peninsula. RVFV is a
member of the Phlebovirus genus, 1 of the 5 genera in the family Bunyaviridae. It
is transmitted by many species of mosquito of the genera Culex and Aedes, in which
transovarial passage can occur. Mosquitoes, once infected, remain so, and in them,
the virus is not pathogenic. High levels of viremia occur in sheep and cattle and
are maintained for up to 5 days. During epizootics, the virus may be spread by fomites,
aerosols, and mechanically by other biting insects.
In endemic situations, the disease in adults is usually mild, but in epidemics in
sheep and goats, severe illness occurs with fever, mucopurulent nasal discharge, and
dysentery. The mortality rate is then very high, reaching 90-100% in lambs and 10-30%
in adults. The disease in cattle is less severe, but pregnant animals abort, and the
mortality rate in adult animals is 5-10%, whereas in calves it may reach 70%.
As in Wesselsbron disease, the gross postmortem picture is dominated by widespread
hemorrhage, ranging from serosal petechiae to severe gastrointestinal bleeding. The
liver in the acute cases in neonatal lambs is similar to that in cases of Wesselsbron
disease, being yellow, swollen, soft, and patchily congested or hemorrhagic. In older
animals and in less acute cases, however, the liver tends to be darker and shows scattered
pale foci of necrosis 1-2 mm in diameter. There may be fibrinous perihepatitis, edema
of the gallbladder wall, and moderate, blood-tinged ascites. Experimental infection
of calves produced encephalomyelitis in an animal that survived the initial viremic
stage.
Within 12 hours of experimental infection of lambs, there are randomly distributed
foci of hepatocellular necrosis in the liver. These foci include aggregates of inflammatory
cells and prominent apoptotic bodies and initially involve about a half dozen hepatocytes
(eFig. 2-18). Within a few hours, however, these primary foci enlarge and may become
almost confluent. In the meantime, the remaining parenchyma may rapidly undergo necrosis
that spares only a small rim of periportal hepatocytes. In naturally infected calves,
the primary foci of necrosis undergo lysis more rapidly than the surrounding parenchyma;
these foci thus have a striking “washed-out” appearance. Where the expanding foci
of necrosis include portal triads, there may follow fibrinous vasculitis and thrombosis.
Fibrin deposition in sinusoids is common, and so is mineralization of necrotic hepatocytes.
Cholestasis is apparent in sections but is not a prominent feature.
eFigure 2-18
Focal necrosis with hemorrhage in Rift Valley fever in a lamb.
(Courtesy S. Youssef.)
Eosinophilic intranuclear inclusion bodies, often elongated, are sometimes seen in
degenerate hepatocytes; there is associated nuclear vesiculation and chromatin margination.
There is necrosis in germinal centers of lymphoid follicles in lymph nodes and spleen
similar to that seen in Wesselsbron disease. Renal glomerular hypercellularity and
necrosis have been described in the experimental disease. By immunohistochemistry,
RVFV antigen can be detected in focal areas in cytoplasm of degenerating and necrotic
hepatocytes but not in nonparenchymal cells.
Ultrastructural studies have shown condensation of degenerate hepatocytes, abundant
apoptosis, and the presence of membrane-bound fragments of hepatocellular cytoplasm,
but sinusoidal lining cells are not notably damaged; the Kupffer cells instead participate
in the uptake of the necrotic and apoptotic hepatocytes. There is abundant fibrin
in sinusoids in the vicinity of the primary foci of necrosis. The intranuclear inclusions
are composed not of recognizable viral particles but of filaments. Virus is occasionally
discernible in the cytoplasm, associated with tubular membranes.
It seems that the primary infection of the liver produces the primary necrotic foci,
from which more virus spreads to damage neighboring parenchyma; the virus clearly
has a marked preference for hepatocytes. The hemorrhagic component of the syndrome
is probably related to consumption of clotting factors; there is no direct evidence
for the endothelial damage seen in infectious canine hepatitis.
In summary, Rift Valley fever is characterized by obvious focal hepatic necrosis,
on which is superimposed an extensive centrilobular and midzonal necrosis; cholestasis
is not as prominent as it is in Wesselsbron disease. The liver lesions in the latter
disease consist of smaller, randomly distributed foci of hepatocellular necrosis,
more active reaction by the sinusoidal lining cells, and more obvious cholestasis.
Equine serum hepatitis (Theiler's disease)
Equine serum hepatitis is a common cause of acute hepatic failure in horses. The disease
was originally described in 1919 by Arnold Theiler, in horses passively immunized
with equine serum against African horse sickness, and was later recognized in horses
passively immunized against anthrax, tetanus, and equine encephalomyelitis. Various
injectable biologics of equine origin have been associated with the disease, including
Clostridium perfringens toxoids, tetanus antitoxin, and equine herpesviral vaccines
prepared from equine fetal tissue and pregnant mare serum. However, although such
an association holds for the majority of cases, there are many, usually sporadic,
cases in horses that have not received any injections. It is unknown how many exposed
horses get subclinical disease, because the condition is seldom diagnosed before the
onset of hepatic failure. However, some horses do recover after transient illness
with jaundice, and some can survive with residual neurologic problems.
Recent metatranscriptomic analysis of serum from 2 index clinical cases in an outbreak
of serum hepatitis in horses passively immunized against botulism, as well as from
the equine hyperimmune plasma product administered before the outbreak, identified
a previously unknown and highly divergent member of the Flaviviridae family, designated
“
Theiler's disease–associated virus
” (TDAV). In this outbreak, 8 of 22 horses developed serum biochemical evidence of
liver injury, although only the 2 index cases developed clinical signs of acute hepatic
insufficiency. A qRT-PCR–based assay found evidence of TDAV in serum from 15 of 17
horses treated with the botulinum antitoxin, as well as in 1 of the 3 donor horses
used to produce the antitoxin, whereas 20 in-contact untreated horses and 20 additional
horses from a separate premises were assay negative. Viral load did not, however,
appear to be predictive of biochemical or clinical extent of hepatic injury. One of
4 horses experimentally inoculated with the virus developed elevated serum liver enzymes.
Four of the original 17 horses continued to be asymptomatically positive for TDAV
1 year later, demonstrating chronic infection, whereas horses that tested negative
initially continued to test negative at 1 year, suggesting that horizontal transmission
is inefficient. Although Koch's postulates remain to be fulfilled, TDAV appears to
be a plausible candidate viral cause of this disease. Further research is necessary
to establish the precise role of this virus in the disease.
The incubation period of disease is typically 42-60 days, sometimes up to 90 days.
Onset of the typical clinical syndrome is sudden, with death occurring in 6-24 hours.
Clinically, there is lethargy; jaundice; photosensitivity; hyperexcitability, often
with mania; continuous walking and pushing; apparent blindness; and ataxia. Death
occurs suddenly without a period of prostration.
At autopsy, icterus is present, with moderate ascites; the spleen is normal or congested,
and there may be petechial hemorrhages on serous membranes and renal cortices, and
some congestion of the intestine with hemorrhage into its lumen. Grossly, the liver
usually appears atrophic, small, and flabby because of the acute loss of many hepatocytes
(Fig. 2-72A
). The liver may be stained by bile pigments, and its surface mottled with a few strands
of fresh fibrin. The mottling is more evident on the cut surface, which may be severely
congested with an apparent zonal pattern, and sometimes fatty (Fig. 2-72B).
Figure 2-72
Equine serum hepatitis (Theiler's disease). A. Small, limp and flaccid “dishrag” liver.
B. The reticular pattern in this slice of liver suggests zonal necrosis.
Microscopically, the hepatic lesion is considerably older than the clinical course
would suggest. There may be a few surviving swollen and vacuolated periportal hepatocytes
or sometimes complete depletion of parenchymal cells in the section. In less-affected
regions in animals with adequate dietary or adipose reserves to mobilize, there is
severe macrovesicular fatty change in most remaining hepatocytes. Acute necrosis is
typically not seen, and there is no significant hemorrhage. Variable numbers of apoptotic
hepatocytes are expected. In the periphery of the acini, severely ballooned cells
undergo dissolution to leave scattered fatty cysts, but most of them disappear to
leave either sinusoids that are dilated and filled with blood or a condensed and distorted
reticulin framework. Extensive deposits of bile pigments are present in Kupffer cells
and hepatocytes. Leukocytes, including lymphocytes, plasma cells, histiocytes, and
a few neutrophils, may infiltrate diffusely, but not in large numbers. There is diffuse
but very slight fibroplasia, especially in the portal units. In some livers, there
is a ductular reaction, with small irregular clusters of proliferating ductular cells
evident in the portal areas.
Further reading
Baba SS, et al. Wesselsbron virus antibody in domestic animals in Nigeria: retrospective
and prospective studies. New Microbiol 1995;18:151-162.
Boomkens SY, et al. Hepatitis with special reference to dogs. A review on the pathogenesis
and infectious etiologies, including unpublished results of recent own studies. Vet
Q 2004;26:107-114.
Carmichael LE. The pathogenesis of ocular lesions of infectious canine hepatitis.
I. Pathology and virological observations. Pathol Vet 1964;1:73-95.
Carmichael LE. The pathogenesis of ocular lesions of infectious canine hepatitis.
II. Experimental ocular hypersensitivity produced by the virus. Pathol Vet 1965;2:344-359.
Chandriani S, et al. Identification of a previously undescribed divergent virus from
the Flaviviridae family in an outbreak of equine serum hepatitis. Proc Natl Acad Sci
U S A 2013;110:E1407-E1415.
Chouinard L, et al. Use of polymerase chain reaction and immunohistochemistry for
detection of canine adenovirus type I in formalin-fixed, paraffin-embedded liver of
dogs with chronic hepatitis or cirrhosis. J Vet Diagn Invest 1998;10:320-325.
Coetzer JAW. The pathology of Rift Valley fever. I. Lesions occurring in natural cases
in new-born lambs. II. Lesions occurring in field cases in adult cattle, calves and
aborted foetuses. Onderstepoort J Vet Res 1977;44:205-212. 1982;49:11-17.
Coetzer JAW, et al. Wesselsbron disease: pathological, haematological and clinical
studies in natural cases and experimentally infected new-born lambs. Onderstepoort
J Vet Res 1978;45:93-106.
Coetzer JAW, Ishak KG. Sequential development of the liver lesions in new-born lambs
infected with Rift Valley fever virus. I. Macroscopic and microscopic pathology. II.
Ultrastructural findings. Onderstepoort J Vet Res 1982;49:103-108, 109-122.
Coyne KP, et al. Lethal outbreak of disease associated with feline calicivirus infection
in cats. Vet Rec 2006;158:544-550.
Fagbo SF. The evolving transmission pattern of Rift Valley fever in the Arabian Peninsula.
Ann N Y Acad Sci 2002;969:201-204.
Gadsden BJ, et al. Fatal Canid herpesvirus 1 infection in an adult dog. J Vet Diagn
Invest 2012;24:604-607.
Halbur PG, et al. Comparative pathogenesis of infection of pigs with hepatitis E viruses
recovered from a pig and a human. J Clin Microbiol 2001;39:918-923.
Herder V, et al. Poxvirus infection in a cat with presumptive human transmission.
Vet Dermatol 2011;22:220-224.
Hoover EA, Griesemener RA. Experimental feline herpesvirus infection in the pregnant
cat. Am J Pathol 1971;65:173-188.
Ikegami T, Makino S. The pathogenesis of Rift Valley fever. Viruses 2011;3:493-519.
Kasorndorkbua C, et al. Experimental infection of pregnant gilts with swine hepatitis
E virus. Can J Vet Res 2008;67:303-306.
Klopfleisch R, et al. Distribution of lesions and antigen of highly pathogenic avian
influenza virus A/Swan/Germany/R65/06 (H5N1) in domestic cats after presumptive infection
by wild birds. Vet Pathol 2007;44:261-268.
Marschall J, Hartmann K. Avian influenza A H5N1 infections in cats. J Fel Med Surg
2008;10:359-365.
Moeller RB, et al. Systemic Bovine herpesvirus 1 infections in neonatal dairy calves.
J Vet Diagn Invest 2013;25:136-141.
Odendaal L, et al. Sensitivity and specificity of real-time reverse transcription
polymerase chain reaction, histopathology, and immunohistochemical labeling for the
detection of Rift Valley fever virus in naturally infected cattle and sheep. J Vet
Diagn Invest 2014;26:49-60.
Pesavento PA, et al. Pathologic, immunohistochemical, and electron microscopic findings
in naturally occurring virulent systemic feline calicivirus infection in cats. Vet
Pathol 2004;41:257-263.
Resendes AR, et al. Apoptosis in postweaning multisystemic wasting syndrome (PMWS)
hepatitis in pigs naturally infected with porcine circovirus type 2 (PCV2). Vet J
2011;189:72-76.
Rippy MK, et al. Rift Valley fever virus-induced encephalomyelitis and hepatitis in
calves. Vet Pathol 1992;29:495-502.
Rosell C, et al. Hepatitis and staging of hepatic damage in pigs naturally infected
with porcine circovirus type 2. Vet Pathol 2000;37:687-692.
Schulze C, Baumgärtner W. Nested polymerase chain reaction and in situ hybridization
for diagnosis of canine herpesvirus infection in puppies. Vet Pathol 1998;35:209-217.
Sinha A, et al. Singular PCV2a or PCV2b infection results in apoptosis of hepatocytes
in clinically affected gnotobiotic pigs. Res Vet Sci 2012;92:151-156.
van der Lugt JJ, et al. The diagnosis of Wesselsbron disease in a new-born lamb by
immunohistochemical staining of virus antigen. Onderstepoort J Vet Res 1995;62:143-146.
Woods LW, et al. Cholangiohepatitis associated with adenovirus-like particles in a
pygmy goat. J Vet Diagn Invest 1991;3:89-92.
Bacterial infections
Bacterial hepatitis is common, but, with a few important exceptions, is usually focally
distributed and of little clinical significance. Bacteria may gain entrance to the
liver in various ways: by direct implantation, for example, by foreign-body penetration
from the reticulum; by invasion of the capsule from an adjacent focus of suppurative
peritonitis; hematogenously via the hepatic artery or portal and umbilical veins;
or via the bile ducts.
Excepting peracute septicemias, there are few specific bacterial infections that have
a sustained or repeated bacteremic phase without producing hepatic lesions. There
are, in addition, many cases of nonspecific bacteremia, especially those originating
in the drainage field of the portal vein, in which focal hepatitis occurs. Because
their differential diagnosis is of some importance, it is probably useful to list
here those specific bacterial diseases in which focal hepatitis is expected or characteristic,
but not constant. The specific infections may occur as fetal or perinatal infections.
The list includes Listeria monocytogenes in fetal and neonatal lambs, calves, foals,
and piglets (Fig. 2-73
); Campylobacter fetus in fetal and neonatal lambs (Fig. 2-74
); Actinobacillus equuli in foals; A. suis in pigs; Yersinia pseudotuberculosis in
lambs and occasionally in dogs and cats; Francisella (Yersinia) tularensis in lambs
and cats; Mannheimia haemolytica and Histophilus somni (Haemophilus agni) in lambs;
Salmonella spp. in all hosts; Clostridium piliforme (Tyzzer's disease) in foals and
dogs; Nocardia asteroides in dogs; and the mycobacteria in all hosts (Figure 2-75,
Figure 2-76
).
Figure 2-73
Multifocal hepatitis caused by
Listeria monocytogenes
infection in a calf.
(Courtesy J.L. Caswell.)
Figure 2-74
Campylobacter fetus
infection in an ovine fetus.
(Courtesy P. Stromberg.)
Figure 2-75
Tuberculosis in a bovine liver.
(Courtesy University of Guelph.)
Figure 2-76
Hepatic mycobacteriosis in a Schnauzer dog A. H&E. B. Acid-fast stain.
(Courtesy J.L. Caswell.)
Hepatic abscess
Hepatic abscesses, quite apart from the lesions of the specific infections just given,
are common, especially in cattle. They may arise by direct implantation of a foreign
body from the reticulum or by direct invasion of the capsule from a suppurative lesion
of traumatic reticulitis and may be single or multiple, but in either case, they are
often preferentially distributed to the left lobe. They may be hematogenous from portal
vein emboli, or by direct extension of omphalophlebitis. Arteriogenic abscesses via
the hepatic artery may occur in pyemias but are quite uncommon.
Omphalogenic abscesses are more common in calves than in other species but occur in
all. The bacterial flora is frequently mixed, but Arcanobacterium pyogenes, Fusobacterium
necrophorum, streptococci, and staphylococci usually predominate. Hepatic abscesses
are not inevitable sequelae to omphalitis or even to omphalophlebitis, but they do
not develop from navel infections in the absence of omphalophlebitis. As there is
no flow of blood in these vessels after birth, involvement of the liver is by direct
growth along the physiologic thrombus. Omphalophlebitis can be quite severe without
extension to the liver. Hepatic abscesses of omphalogenic origin are often restricted
to the left lobe (Fig. 2-77
), but they may be restricted to the right or be generalized in their distribution.
Figure 2-77
Omphalophlebitis with miliary metastatic abscesses in the left lobe of the liver in
a calf.
Hepatic abscesses are also common and of economic importance in feedlot cattle. They
are usually found at slaughter but, when numerous, may be fatal after a few days of
vague digestive illness. Their pathogenesis and character are discussed with rumenitis,
to which they are a sequel (see Vol. 2, Alimentary system and peritoneum). Liver abscesses
in feedlot sheep likely have a similar pathogenesis, with F. necrophorum as the primary
isolate. A second category includes parasitic granulomas populated by various opportunistic
bacteria.
Hepatic abscesses of biliary origin occur in all animals. They occur in pigs in which
ascarids have migrated into the bile ducts. Cholangitic abscesses in horses, dogs,
and cats are usually caused by enterobacteria as part of a fulminating ascending cholangiohepatitis
that is fatal after a short course.
The sequelae of hepatic abscessation are variable. Usually, they are insignificant
and asymptomatic. Sterilization of the focus with either resorption and complete healing
or encapsulation is common. Those near the surface of the liver regularly produce
fibrinous and then fibrous inflammation of the capsule and adhesion to adjacent viscera.
They seldom perforate the capsule but do commonly break into hepatic veins to produce
any one or a combination of thrombophlebitis of the vena cava, endocarditis, or pulmonary
abscesses or embolism. Acute extension of a hepatic abscess into the major hepatic
vein can lead to pulmonary embolism that can be acutely fatal. In adults, death may
occur if the hepatic abscesses are multiple and fresh, and especially if they are
necrobacillary in origin; death is probably the result of toxemia.
Hepatic necrobacillosis
Occasionally,
F. necrophorum
infection of the liver is observed following omphalophlebitis in lambs and calves,
or as a complication of rumenitis in adult cattle. In feedlot cattle, both F. necrophorum
subsp. necrophorum (biotype A) and subsp. funduliforme (biotype B) have been isolated.
The hepatic lesions are multiple slightly elevated, rounded, dry areas of coagulative
necrosis, sometimes a few centimeters in diameter (eFig. 2-19) and surrounded by a
zone of intense hyperemia. Affected neonates seldom live long enough for the necrotic
foci to liquefy and assume the appearance of ordinary abscesses, but this may be seen
in adult cattle. The histologic appearance of the foci in the stage of coagulative
necrosis is quite characteristic. The necrotic amorphous central area is bordered
by a zone of wholesale destruction of leukocytes, whose nuclear chromatin is dissipated
in a finely divided form, and among which the filamentous fusobacteria are mostly
concentrated. Outside this zone, there is severe hyperemia and hemorrhage, and thrombosis
of local vessels is common. The lesion in neonatal lambs is to be distinguished from
that caused by Campylobacter fetus.
eFigure 2-19
Hepatic necrobacillosis (Fusobacterium necrophorum infection) in an ox. Pale slightly
raised areas of dry coagulative necrosis bordered by acute inflammation.
(Courtesy University of Guelph.)
The pathogenic mechanisms of F. necrophorum involve various toxins, in particular,
a high-molecular-weight leukotoxin specifically toxic to ruminant neutrophils. This
unique toxin activates neutrophils and induces their apoptosis, consistent with the
remarkable abscess-inducing propensity of F. necrophorum in ruminants. However, other
toxins, endotoxic lipopolysaccharide (LPS), and hemagglutinins are also implicated
as virulence factors. Mixed infections are frequent, and synergism between F. necrophorum
and other pathogens, such as Arcanobacterium pyogenes, may also play a role in the
pathogenesis of liver necrosis and abscessation.
Necrotic hepatitis (black disease)
Organisms of the genus Clostridium are notably circuitous in their means of producing
disease. This is true of
C. novyi; type B strains, which produce potent exotoxins and are the cause of black
disease (infectious necrotic hepatitis). The alpha toxin of C. novyi is related to
the large clostridial cytotoxins produced by C. difficile and C. sordelli. These toxins
enter cells by receptor-mediated endocytosis and inhibit ras and rho guanosine triphosphatases
by glycosylation. The beta toxin is a necrotizing and hemolytic phospholipase C (lecithinase).
Black disease occurs in nonimmune animals when these exotoxins are released by C.
novyi within an anaerobic focus in the liver. These anaerobic sites, which provide
a suitable environment for germination of C. novyi spores, are most commonly a result
of migrating liver flukes.
C. novyi is widely distributed in soil, and the spores are continually ingested by
grazing animals in areas where black disease occurs. Some spores cross the mucous
membranes, probably in phagocytes, and remain as latent infections in macrophages,
mainly in the liver, spleen, and bone marrow. The duration of latency in tissue is
not known, but it can be many months. In endemic areas, many healthy sheep, cattle,
and dogs harbor latent infections in their livers. Black disease is principally an
acutely fatal disease of sheep in regions where the inciting helminths are endemic.
The disease is most commonly initiated by migrating larvae of the common liver fluke
Fasciola hepatica. In Bessarabia and France, it is endemically related to the distribution
of Dicrocoelium dendriticum, the lancet liver fluke. Sporadic cases may be related
to Cysticercus tenuicollis infection or may be idiopathic.
Deaths in sheep from black disease occur rapidly and usually without warning signs.
Illness, if observed, is brief and characterized by reluctance to move, drowsiness,
rapid respiration, and quiet subsidence. Affected animals are usually in good nutritional
condition. Postmortem decomposition occurs rapidly. The name of the disease is derived
from the appearance of flayed skins, the dark coloration being caused by an unusual
degree of subcutaneous venous congestion. Frequently, there is edema of the sternal
subcutis, and airways contain stable foam. Serous cavities contain an abundance of
fluid that clots on exposure to air. The fluid is usually straw colored, but that
in the abdomen may be tinged with blood. The volume of fluid in the abdomen and thorax
may vary from about 50 mL to 1.5 L. The pericardial sac is distended with similar
fluid in amounts up to ~300 mL. Subendocardial hemorrhages in the left ventricle are
almost constant. Patchy areas of congestion and hemorrhage may be present in the pyloric
part of the abomasum and in the small intestine.
The typical and diagnostic lesions occur in the liver and are always present. They
are usually clearly evident on the capsular surface, the diaphragmatic surface especially,
but the organ may have to be sliced carefully to find them. The liver will be the
seat of either the acute traumatic hemorrhagic lesions of acute fascioliasis, the
cholangiohepatitis of the chronic disease, or both. The lesion of black disease, and
occasionally there are several, is a yellow-white area of necrosis 2-3 cm in diameter,
surrounded by a broad zone of intense hyperemia, roughly circular in outline, and
extending hemispherically into the substance of the organ. There may be a coagulum
of fibrin on the capsular surface overlying the necrotic area. Occasionally, the essential
lesions are rectilinear in shape or very irregular. The lesions appear homogeneous
on the cut surface, but some contain poorly defined centers of soft or caseous material.
The histologic evolution of the hepatic lesions begins with the necrotic and hemorrhagic
tracts caused by wandering immature flukes. These are sinuous tunnels ~0.5 cm in diameter
that contain blood, necrotic hepatic cells, and the leukocytes, chiefly eosinophils,
attracted by the flukes. About the tunnels is a narrow zone of coagulative necrosis,
also produced by the flukes. As usual, the necrotic tissue is demarcated by a thin
zone of scavenger cells, chiefly neutrophils. If latent spores are present in the
necrotic areas, they quickly vegetate and are visible in sections as large, gram-positive
bacilli. In nonimmune animals, the vegetative organisms elaborate exotoxins that cause
necrosis of the surrounding tissue, including the eosinophils of the fluke tunnel.
As the area of necrosis expands, the bacterial proliferation keeps pace so that bacilli
can be found in all parts of the necrotic focus but not in the surrounding viable
tissue. Usually, bacteria are concentrated at the advancing margin of the lesion,
just inside a zone of infiltrated neutrophils. At about the time of death and immediately
afterward, the bacilli scatter in the liver and to other organs.
Bacillary hemoglobinuria
Bacillary hemoglobinuria is a counterpart of black disease. The cause is
Clostridium haemolyticum,
which is closely related to C. novyi. Both species produce the beta toxin, a necrotizing
and hemolytic lecithinase (phospholipase C). The pathogenesis of the 2 diseases is
comparable, as both depend on a focus of hepatic injury within which latent spores
can germinate. Bacillary hemoglobinuria as an endemic disease exists only in areas
where Fasciola hepatica abounds, and it is probable that flukes are the primary cause
of the initiating lesion. The disease does occur sporadically where there are no flukes
and may be prompted by other parasites or other diverse focal lesions, which are smudged
out in the expanding areas of necrosis. There is scant information on the ecology
of the organism, but it is clear that it has its own environmental requirements, and
the disease will not persist in areas where these requirements are not met. The spores
will remain in the livers of cattle for several months after removal from pastures
where the disease is endemic. Spores may persist in the bones of cadavers for 2 years.
Spores of this and other sporulating anaerobes can frequently be demonstrated in the
liver, where they are probably retained in Kupffer cells.
Bacillary hemoglobinuria occurs in cattle and sheep. It is characterized clinically
by intravascular hemolysis with anemia and hemoglobinuria, but, perhaps reflecting
variety in exotoxins between strains of the organism, hemolysis may not be a feature.
The essential lesion is hepatic and similar to that of black disease but is much larger
and usually single (Fig. 2-78
). It has been described as an infarct secondary to portal thrombosis, and although
this may occur in isolated cases, it is scarcely a creditable pathogenesis for a disease
of endemic occurrence. Thrombosis does occur in the affected areas but can be a result,
rather than a cause, of the initial lesion and is found more frequently in the hepatic
venules than in branches of the portal vein. There is severe anemia, the kidneys are
speckled red or brown by hemoglobin, and the urine is of port-wine color. Peritoneal
vessels are injected, and in some cases, there is severe, dry, fibrinohemorrhagic
peritonitis.
Figure 2-78
Large focus of hepatic necrosis in an ox with bacillary hemoglobinuria.
(Courtesy University of Guelph.)
Clostridium piliforme infection
Clostridium piliforme (formerly Bacillus piliformis) infection has long been known
as Tyzzer's disease, a cause of severe losses in laboratory rodents; however, it has
also been reported in foals, calves, dogs, and cats. Although the disease is probably
initiated by an intestinal infection, lesions in the gut are less specific and constant
than those in the liver, which consist of focal hepatitis and necrosis.
Affected foals usually die between the ages of 1-4 weeks; often they are found dead
after a short illness. The liver shows pale foci up to a few millimeters across (Fig.
2-79
); these are represented microscopically by randomly distributed foci of coagulative
necrosis with moderate neutrophilic infiltrate (Fig. 2-80A
). This lesion is not diagnostic in itself; its specificity depends on the presence
of the causal organism in hepatocytes in the periphery of the necrotic zones. At present,
C. piliforme can only be isolated with difficulty on artificial media, so diagnosis
is usually based on demonstration of the large, long bacilli in the cytoplasm of degenerate
and also otherwise apparently normal hepatocytes at the periphery of the necrotic
zones. The organisms are gram negative and are best delineated with silver impregnation
techniques, such as that of Warthin-Starry, but they may be seen with routine stains,
such as Giemsa, particularly when the material is fresh. Differentiation from other
organisms, including postmortem saprophytes, can be achieved by immunohistochemistry
or immunofluorescence. The bacilli tend to lie in sheaves or bundles (Fig. 2-80B).
There may also be colitis sufficiently severe to cause diarrhea, but not as severe
as that seen in rabbits with this disease.
Figure 2-79
Foal liver with multifocal hepatitis in
Clostridium piliforme
infection (Tyzzer's disease).
Figure 2-80
Tyzzer's disease in a foal. A. Foci of necrosis and suppurative inflammation. B.
Clostridium piliforme bacilli in bundles in hepatocytes at margin of lesion. Warthin-Starry
silver stain.
Fewer cases of Tyzzer's disease have been reported in dogs and cats. The liver lesions
are essentially the same as those in foals and rodents, and there is also enteritis
or enterocolitis. Immunodeficiency predisposes to the disease because it occurs sporadically
in dogs that have undergone immunosuppressive or anticancer therapy. Such cases may
be complicated by concurrent viral, mycotic, or protozoal infections.
Leptospirosis
Leptospirosis is a systemic disease characterized by acute jaundice, cholestatic liver
injury, and renal failure in dogs, and is discussed in more detail in Vol. 2, Urinary
system. The hepatic lesions described following acute experimental infection of dogs
with Leptospira kirschneri serovar grippotyphosa include mixed perivascular periportal
infiltrates of neutrophils, lymphocytes, and plasma cells, with mild hepatic lipidosis,
dissociation of hepatocytes, and intracanalicular bile plugs evident by day 12 postinfection,
along with increased hepatocellular mitotic activity. Clinical icterus has been attributed
to cholestasis caused by dissociation of hepatocytes (eFig. 2-20). Dogs experimentally
infected with Leptospira interrogans serovar pomona developed pulmonary and renal
petechial hemorrhages, and friable livers with multifocal 1-2 mm raised white foci
corresponding to periportal inflammatory infiltrates of lymphocytes, plasma cells,
neutrophils, and macrophages; small foci of hepatic necrosis; and bile plugs in canaliculi,
in addition to renal, pulmonary, and cardiac lesions. Organisms were identified by
immunohistochemistry at the brush borders of renal proximal convoluted tubules as
well as at the luminal surface of bile duct epithelium. In a retrospective study of
dogs with supportive clinical signs and microscopic agglutination test titers of ≥320
for one or more serovars tested, including autumnalis, bratislava, canicola, grippotyphosa,
icterohaemorrhagiae, and pomona, histologic lesions in the liver were subtle, with
sinusoidal neutrophil margination, Kupffer cell hypertrophy, and low levels of hepatocellular
single-cell necrosis and mitoses. Some livers had diffuse interstitial lymphocytic
hepatitis, with mitotic figures, anisokaryosis, binucleation, and some degree of lobular
collapse. Chronic hepatitis has also been experimentally produced by leptospiral infection
in dogs; however, clinical cases are rarely documented.
eFigure 2-20
Leptospirosis with dissociation of hepatocytes in a dog.
Other bacteria
A single case of clinical disease associated with
Helicobacter canis
has been reported in a 2-month-old puppy with peracute disease causing weakness and
vomiting before death. Multiple coalescing yellow foci in the liver up to 1.5 cm in
diameter consisted of hepatocellular coagulative necrosis with infiltrating mononuclear
cells and neutrophils. Spiral bacteria were visualized by Warthin-Starry silver stains
in area of necrosis, within bile canaliculi, and occasionally in bile duct lumens.
This organism has previously been identified in the blood of diarrheic children, and
in the feces of 4% of dogs in an epidemiologic study examining the incidence of Campylobacter-like
organisms in 1,000 dogs.
Bartonella
spp. infection has been associated with a wide variety of granulomatous syndromes
and peliosis hepatis in humans, often in association with immunosuppression. Bartonella
henselae and B. clarridgeae are associated with self-limiting illness and persistent
intravascular infection in cats; mild lymphocytic cholangitis/pericholangitis and
lymphocytic hepatitis have been reported with experimental infection of cats with
B. henselae. B. henselae has been identified in liver tissue by PCR from a single
case of peliosis hepatis in a Golden Retriever and granulomatous hepatitis in a Basset
Hound, whereas B. clarridgeae DNA was identified in liver tissue from a Doberman Pinscher
with histologic lesions of Doberman hepatitis, however, the extent to which infection
induces disease in dogs is currently unknown.
Further reading
Adamus C, et al. Chronic hepatitis associated with leptospiral infection in vaccinated
beagles. J Comp Pathol 1997;117:311-328.
Baldwin CJ, et al. Acute tularemia in three domestic cats. J Am Vet Med Assoc 1991;199:1602-1605.
Farrar ET, et al. Hepatic abscesses in dogs: 14 cases (1982-1994). J Am Vet Med Assoc
1996;208:243-247.
Fox JG, et al. Helicobacter canis isolated from a dog liver with multifocal necrotizing
hepatitis. J Clin Microbiol 1996;34:2479-2482.
Gillespie TN, et al. Detection of Bartonella henselae and Bartonella clarridgeiae
DNA in hepatic specimens from two dogs with hepatic disease. J Am Vet Med Assoc 2003;222:47-51.
Greenlee JJ, et al. Clinical and pathologic comparison of acute leptospirosis in dogs
caused by two strains of Leptospira kirschneri serovar grippotyphosa. Am J Vet Res
2004;65:1100-1107.
Greenlee JJ, et al. Experimental canine leptospirosis caused by Leptospira interrogans
serovars pomona and bratislava. Am J Vet Res 2005;66:1816-1822.
Ikegami T, et al. Naturally occurring Tyzzer's disease in a calf. Vet Pathol 1999;36:253-255.
Iwanaka M, et al. Tyzzer's disease complicated with distemper in a puppy. J Vet Med
Sci 1993;55:337-339.
Kordick DL, et al. Clinical and pathologic evaluation of chronic Bartonella henselae
or Bartonella clarridgeae infection in cats. J Clin Microbiol 1999;37:1536-1547.
Lechtenberg KF, et al. Bacteriologic and histologic studies of hepatic abscesses in
cattle. Am J Vet Res 1988;49:58-62.
Narayanan S, et al. Fusobacterium necrophorum leukotoxin induces activation and apoptosis
of bovine leukocytes. Infect Immun 2002;70:4609-4620.
O'Toole D, et al. Tularemia in range sheep: an overlooked syndrome? J Vet Diagn Invest
2008;20:508-513.
Paar M, et al. Infection with Bacillus piliformis (Tyzzer's disease) in foals. Schweiz
Arch Tierheilkd 1993;135:79-88.
Prescott JF, et al. Resurgence of leptospirosis in dogs in Ontario: recent findings.
Can Vet J 2002;43:955-961.
Rissi DR, Brown CA. Diagnostic features in 10 naturally occurring cases of acute fatal
canine leptospirosis. J Vet Diagn Invest 2014;26:799-804.
Scanlan CM, Edwards JF. Bacteriologic and pathologic studies of hepatic lesions in
sheep. Am J Vet Res 1990;51:363-366.
Tadepalli S, et al. Fusobacterium necrophorum: a ruminal bacterium that invades liver
to cause abscesses in cattle. Anaerobe 2009;15:36-43.
Tan ZL, et al. Fusobacterium necrophorum infections: virulence factors, pathogenic
mechanism and control measures. Vet Res Commun 1996;20:113-140.
Warner SL, et al. Clinical, pathological, and genetic characterization of Listeria
monocytogenes causing sepsis and necrotizing typhlocolitis and hepatitis in a foal.
J Vet Diagn Invest 2012;24:581-586.
Helminthic infections
Various helminths, including cestodes, nematodes, and trematodes, as well as the larval
pentastome Linguatula serrata, can produce inflammation of liver and bile ducts. Those
parasites that have the biliary system as their final habitat will be discussed in
detail later. The others produce hepatic lesions in the course of their natural or
accidental migrations, and are discussed under the organ (for most of them, the gut)
that is their final habitat. It is useful, however, to describe briefly here the lesions
produced by larvae in transit.
The initial lesion produced by wandering larvae is traumatic. Sinuous tunnels permeate
the parenchyma and often breach the capsule. In the tunnels, there are free red cells,
degenerating hepatocytes, and leukocytes, chiefly eosinophils, which react to the
parasites. Bordering the tunnel is a narrow zone of coagulative necrosis of parenchyma
with neutrophils at its margin. Eosinophils also infiltrate the portal triads. The
necrotic parasitic tracts heal by scarification, and the fibroblastic tissue, infiltrated
with eosinophils, is eventually incorporated into the portal units. Most larvae escape
from the liver, but some eventually become encapsulated in the liver in abscesses
containing numerous eosinophils. The abscesses may caseate and come to resemble tubercles,
and eventually many are heavily mineralized to form permanent pearly nodules. In sheep,
the most common cause of this type of hepatitis (aside from liver fluke) is
Cysticercus tenuicollis
in its wandering phase. Lambs may die of severe hemorrhagic hepatitis (Fig. 2-81
) caused by very heavy infections of this parasite, and in pigs, an aberrant host,
C. tenuicollis can produce a very intense inflammatory reaction.
Figure 2-81
Hemorrhagic subcapsular migration tracks caused by Cysticercus tenuicollis in a lamb.
(Courtesy A. Rehmtulla.)
In pigs, larvae of
Ascaris suum
and
Stephanurus dentatus
produce similar but distinct patterns of focal interstitial hepatitis. The ascarids
produce their distinctive accentuation of the stroma (“milk spots”) (Fig. 2-82
) when quite small larvae are immobilized by the host's inflammatory reaction; thus
the foci are relatively small. The fibrotic lesion produced by S. dentatus larvae,
on the other hand, is less focal and more in the nature of a track, and there are
usually small, inflamed, capsular craters where the larvae have emerged from the liver
to migrate to their preferred perirenal site. There will be obvious portal phlebitis
at the hepatic hilus when infection by S. dentatus has been by the oral route, and
in these livers, the parenchymal lesion is more severe in this vicinity.
Figure 2-82
Multifocal interstitial hepatitis (“milk-spot liver”) caused by Ascaris suum migration
in a pig.
Migration tracks left by larval strongyles can be found under the liver capsule in
young horses, and historically, these have been thought to relate to the dense, discrete,
capsular fibrous tags and plaques that are almost universally found on the diaphragmatic
surface of the liver of mature horses. However, these hepatic fibrotic lesions remain
common in horses even after the widespread use of effective anthelmintics, and their
precise etiology remains unknown (Fig. 2-83
).
Figure 2-83
Fibrous tags on the diaphragmatic surface of an equine liver.
(Courtesy University of Guelph.)
Parasites can produce hepatic lesions by other means. The hydatid intermediate stages
of Echinococcus encyst in the liver and may destroy much of it; the larvae of Ascaris
suum in cattle add to the usual insult by causing portal phlebitis and small areas
of infarction; the adults of Ascaris in all species, but especially in pigs, may migrate
into the bile ducts; and the eggs of schistosomes enter in the portal blood to lodge
in the intrahepatic portal vessels and provoke granulomatous inflammation.
Cestodes
Stilesia hepatica
and
Thysanosoma actinioides,
the “fringed tapeworm,” are the only cestodes that inhabit the bile ducts. They are
parasites of ruminants, Stilesia occurring in Africa, Thysanosoma in North and South
America. The life cycles of the parasites are not completely known but may involve
oribatid mites as intermediate hosts. T. actinioides may also be found in the pancreatic
ducts and small intestine. Usually, the infestations are light but, even when heavy,
are not of much significance (eFig. 2-21). Very heavy infestations by S. hepatica
occur without signs of illness, although the bile ducts may be nearly occluded, slightly
thickened, and dilated. Saccular dilations of the ducts may occur and be filled with
worms. The fringed tapeworm is perhaps more pathogenic, and unthriftiness may accompany
heavy infestations.
eFigure 2-21
Fringed tapeworms Thysanosoma actinioides
in the bile ducts of a sheep.
(Courtesy A. Reid.)
Echinococcus granulosus
hydatid cysts occur most commonly in the liver of ruminants in endemic areas (Fig.
2-84
) but have been reported as incidental postmortem findings in the liver of pigs and
horses. The cysticerci and hydatids, which in the intermediate stages invade the liver,
are discussed further in Vol. 2, Alimentary system and peritoneum.
Figure 2-84
Hydatid liver disease (Echinococcus granulosus infestation) in a sheep.
(Courtesy University of Guelph.)
Echinococcus multilocularis
is found in Europe, parts of northern Asia, and North America, where it has a sylvatic
life cycle. Red and arctic foxes are the most important definitive hosts, although
coyotes, raccoon-dogs, and wolves can also be infected. In North America, there are
few documented cases of intestinal infection by E. multilocularis tapeworms in domestic
dogs and even fewer in cats. The adult tapeworm is small, 5 mm, and infective eggs
are shed in the feces to be ingested by the intermediate host, which in temperate
areas are typically various species of rodents, including voles, lemmings, and deer
mice. The hexacanth embryo is released from the egg, travels through the intestinal
wall, and migrates to the liver via the hepatic portal circulation. The metacestode
stage is an alveolar hydatid cyst, composed of numerous small vesicles lined by a
PAS-positive acellular laminated membrane or cuticle layer and a layer of germinal
epithelium from which protoscolices develop. Some infections in dogs do not progress
to form protoscolices, and instead form irregular large cysts with a PAS-positive
laminated membrane (Fig. 2-85
). Additional exogenous budding results in spread of the metacestodes to other internal
locations. The life cycle is completed when the infected intermediate host is consumed
by a definitive host, where the protoscolices attach to the intestinal wall and mature.
Other species can occasionally act as aberrant intermediate hosts, including pigs,
horses, and humans; human alveolar echinococcosis is a serious zoonosis. Dogs can
simultaneously act as definitive and intermediate hosts for this parasite. Cases of
hepatic alveolar hydatid cysts have been reported in dogs from highly endemic areas
in Belgium, Germany, Switzerland, and most recently in Canada (British Columbia, Ontario).
Affected dogs display progressive abdominal enlargement, intermittent inappetance,
and vomiting, and the cysts may be found in the liver, omentum, abdominal cavity,
or lungs.
Figure 2-85
Echinococcus multilocularis cysts with acellular laminated membranes in the liver
of a dog. PAS stain.
(Courtesy A. Brooks.)
Nematodes
Calodium hepaticum (Capillaria hepatica) is the one nematode that in the adult phase
inhabits the liver. It is a slender worm, morphologically resembling the whipworms,
and it lives in the parenchyma rather than in the bile ducts. The usual hosts of the
adult stage are rodents, but sporadic infestations are observed in dogs. These worms
are not highly pathogenic. The adults provoke some traumatic hepatitis, and the eggs,
which are deposited in clusters, provoke the development of localized granulomas.
The eggs are readily recognized by their ovoid shape and polar caps. The granulomas
can be seen through the capsule or in the substance of the liver as yellow streaks
or patches. The eggs cannot escape from the liver unless a predator eats them. Predators,
however, act only as transport hosts, and the ingested eggs are passed in the feces.
Larvae develop in the eggs only in the external environment, and the cycle is completed
when a suitable host eats the mature larvae in the eggs.
Trematodes
Various trematodes (flukes) are parasitic in the livers of animals. They belong to
the families Fasciolidae (Fasciola hepatica, F. gigantica, Fascioloides magna), Dicrocoeliidae
(Dicrocoelium dendriticum, D. hospes, Platynosomum concinnum), Schistosomatidae (Heterobilharzia
americana), and Opisthorchiidae (including Metorchis spp., Opisthorchis felineus,
Clonorchis sinensis, Pseudamphistomum truncatum). The diseases produced are known
collectively as distomiasis.
Fasciola hepatica,
the common liver fluke of sheep and cattle, is the most widespread and important of
the group. Patent infestations can develop in other wild and domestic animals and
in humans. These flukes are leaf shaped and ~2.5 cm long in sheep and slightly larger
in cattle. They are found in the bile ducts. Being hermaphroditic, only one fluke
is necessary to establish a patent infestation, and each adult may produce 20,000
eggs per day. The longevity of the adult flukes is amazing and is potentially as great
as or greater than that of the host; they have been known to survive for 11 years,
and it seems that they can produce eggs all this time. The eggs are eliminated in
bile, and on pasture, in conditions of suitable warmth and moisture, hatch a larva
(miracidium) in ~9 days. If the environmental temperature is low, the incubation period
may be delayed for some months. The miracidium can survive only in moisture. It is
actively motile and penetrates the tissues of the intermediate host, which is an aquatic
snail. Different snails serve this purpose in different countries, but all of them
belong to the genus Lymnaea.
Each miracidium, on penetrating a snail, develops into a mother sporocyst that reproduces,
probably parthenogenetically, giving rise to a small number of the second generation,
the redia. Each redia produces either redia or cercariae, or to the 2 successively.
Cercariae, the larval stage of the third (sexual) generation, first appear 1-2 months
after the miracidium penetrates. Cercariae continue to escape daily for the life of
the snail, but even so, total cercarial production is only 500-1,000. They actively
escape from the snail and are attracted to green plants, where they encyst and become
infective metacercariae in 1 day. These can remain infective for 1 month in summer
and up to 3 months in winter. The developmental events from egg to this stage take
1-2 months under favorable conditions.
Infestation occurs by ingestion. Excystment occurs in the duodenum. The young flukes
penetrate the intestinal wall and cross the peritoneal cavity, attaching here and
there to suck blood and penetrate the liver through its capsule; a few no doubt pass
in the portal vessels or migrate up the bile duct. They wander in the liver for a
month or more before settling down in the bile ducts to mature, which they do in 2-3
months. Some may, by accident, enter the hepatic veins and systemic circulation to
lodge in unusual sites; intrauterine infestations are on record. Lesions caused by
aberrant flukes are quite common in bovine lung. They consist of resilient nodules
just under the pleura of the peripheral parts of the lung. They range in diameter
from ~1 to many centimeters and consist of thinly encapsulated abscesses situated
at the ends of bronchi. The content is slightly mucoid, unevenly coagulated brown
fluid; in some lesions, the reaction is predominantly caseous. The location of the
lesion suggests that it begins as a peripheral bronchiectasis that later becomes sealed
off. The fluke persists in the debris, but is small and hard to find.
The essential lesions produced by F. hepatica occur in the liver and may be described,
first, as those produced by the migratory larvae, and second, as those produced by
the mature flukes in the bile ducts; the two often intermingle (eFig. 2-22). There
is the further incidence of peritonitis, which is produced by the young flukes on
their way to the liver and, perhaps also, by some that break out through the capsule.
eFigure 2-22
Fasciola hepatica infection in a sheep.
(Courtesy J.L. Caswell.)
Usually, there is no obvious reaction to the passage of young flukes through the intestinal
wall and across the peritoneal cavity, except for small hemorrhagic foci on the peritoneum,
where the flukes have been temporarily attached. Few or many parasites may be found
in any ascitic fluid and attached to the peritoneum of the diaphragm and the mesenteries.
When the infestations are heavy and repeated, such as may be observed in sheep, cattle,
and swine, peritonitis occurs. The young flukes at this stage are <1.0 mm long. The
peritonitis may be acute and exudative or chronic and proliferative. It is usually
concentrated on the hepatic capsule, especially its visceral surface, but may be restricted
to the parietal peritoneum or to the visceral peritoneum, including the mesenteries
of the gut. In acute cases, there are fibrinohemorrhagic deposits on the serous surfaces,
and in chronic cases, there may be fibrous tags, with adhesions or a more or less
diffuse thickening by connective tissue. Many young flukes can be found microscopically
in the fibrinous deposits, and in the diffuse peritoneal thickenings, there are tortuous
migration tunnels containing blood, debris, and the young parasites. In cases with
involvement of the visceral peritoneum, young flukes can be found in enlarged mesenteric
lymph nodes.
The acute lesions in the liver caused by the wandering flukes are basically traumatic,
but there is an element of coagulative necrosis, which is possibly related to toxic
excretions of the flukes. The migratory pathways are tortuous tunnels that appear
on cross-section as hemorrhagic foci 2-3 mm in diameter. If a tunnel is followed,
a young fluke <1.0 mm long can be found at the end. When the infestation is heavy,
the liver may appear to be permeated by dark hemorrhagic streaks and foci. Older tunnels
from which the debris has been cleared may appear as light yellow streaks because
of infiltration of eosinophils. Microscopically, fresh tunnels are filled with blood
and degenerate hepatocytes and are soon infiltrated by eosinophils. Later, macrophages
and giant cells arrange themselves about the debris and remove it, and healing occurs
by granulation tissue, which is rich in lymphocytes and eosinophils. In light infestations,
the scars may disappear, but in heavy infestations, they may fuse with each other
and with portal areas to produce moderate irregular fibrosis. There may, as yet, be
no change in the bile ducts. Probably, most of the young flukes reach the bile ducts,
but some do not and become encysted in the parenchyma. One or more flukes may be present
in each cyst, which consists of a connective-tissue capsule and a dirty brown content
of blood, detritus, and excrement from flukes. The cysts ultimately caseate and may
mineralize or are obliterated by fibrous tissue. These cysts are most frequent on
the visceral surface, where they cause bulging of the capsule.
Heavy infestations by immature flukes may cause death at the stage of acute hepatitis.
Such an outcome is not common, but occurs in sheep. It is estimated that 10,000 metacercariae
ingested over a short period are necessary to produce acute death in sheep. Death
may occur suddenly or after a few days of fever, lassitude, inappetance, and abdominal
tenderness. This is also the stage of the parasitism in which black disease occurs.
Mature flukes are present in the larger bile ducts and cause cholangitis. The relative
importance of different factors in their pathogenicity is not known, but they cause
mechanical irritation by the action of their suckers and scales, cause obstruction
of the ducts with some degree of biliary retention, predispose to bacterial infections,
suck blood, and probably produce toxic and irritative metabolic excretions.
The biliary changes occur in all lobes but are usually most severe in the left, and
the right may be moderately hypertrophied. From the hilus, the bile ducts on the visceral
surface stand out as white, firm, branching cords that in extreme instances may be
2 cm in diameter and allow detectable fluctuation over extended segments or in localized
areas of ectasia. This dilation of the ducts in sheep, swine, and horses is largely
mechanical and is caused by distension by masses of flukes and bile. It is permitted
by the relative paucity of new connective-tissue formation in the walls of the ducts
in these species; this in turn is probably related to the rather mild catarrhal type
of inflammation in the lumina of the ducts. In cattle, desquamative and ulcerative
lesions in the large bile ducts are more severe than in other species, and there is
a correspondingly greater proliferation of granulation tissue in and about the walls
of the ducts (Fig. 2-86
). The walls of the ducts in cattle are, in consequence, much thickened, and the lumen
is irregularly stenotic and dilated and lined largely by granulation tissue. This
contributes the typical “pipe stem” appearance to the ducts in cattle; the connective
tissue may be, in addition, mineralized, sometimes so heavily that it cannot be cut
with a knife. The bile ducts contain dirty dark brown fluid of a mucinous or tough
consistency, formed from degenerate floccular bile, pus, desquamated cells and detritus,
clumps of flukes, and small masses of eggs in dark brown granular aggregates (eFig.
2-23).
Figure 2-86
Thickened, dilated bile ducts, caused by
Fasciola hepatica
infection in an ox.
(Courtesy J.L. Caswell.)
eFigure 2-23
Thickened, dilated bile ducts, caused by
Fasciola hepatica
infection in an ox.
(Courtesy J.L. Caswell.)
Although the lesions are most obvious in ducts large enough to contain the flukes,
there is, with time and severe or repeated infestations, progressive inflammation
in the smaller portal units because of direct irritation by the flukes, superimposed
infections, and biliary stasis. The course of events is as described earlier for subacute
and chronic cholangitis. The proliferating connective tissue and bile ductules in
individual portal areas extend to join each other and the scars left over from the
migratory phase, so that inflammatory fibrosis may obliterate parenchyma in many foci.
In such livers, the left lobe, which is the one most severely affected, may be atrophied,
indurated, and irregular.
The development of cholangitis of the degree described depends on long-standing or
heavy infestations. Lesions of lesser severity, or those less fully developed, are
associated with light infestations of short duration. They may then be recognized
only by local dilations of the ducts, or even these may not be readily apparent. In
such mild infestations, the fact of past or present parasitism may only be suggested
by the detection of characteristic black iron-porphyrin pigments, grossly visible
in the hilar nodes. It also contributes to the character of the biliary contents.
Chronic debility with vague digestive disturbances is common in chronic fascioliasis,
and deaths are common among sheep. Clinically and at postmortem, there are, in addition
to the essential lesions, more or less severe anemia, moderate anasarca, and cachexia.
Jaundice is seldom seen.
Fasciola gigantica
displaces F. hepatica as the common liver fluke in many parts of Africa and in nearby
countries, southeast Asia, and the Hawaiian Islands. It is 2 or 3 times as large as
F. hepatica, but its life cycle and pathogenicity are comparable.
Fascioloides magna is the large liver fluke of North America; cervids are the definitive
hosts. It is a parasite of ruminants and lives in the hepatic parenchyma, not in the
bile ducts, although in tolerant hosts (cervids) the cysts in which it localizes communicate
with the bile ducts to provide an exit for ova and excrement. The life cycle of this
parasite generally parallels that of Fasciola hepatica. The young flukes are very
destructive as they wander in the liver. In cattle, they wander briefly, producing
large necrotic tunnels before becoming encysted. The cysts, enclosed by connective
tissue, do not communicate with bile ducts but form permanent enclosures for the flukes,
their excreta, and ova. The cysts, which may be 2-5 cm in diameter, are remarkable
for the large deposits of jet black, sooty iron-porphyrin pigment they contain (eFig.
2-24), and except for the flukes and soft contents, they superficially resemble heavily
pigmented melanotic tumors. Commonly, these flukes pass from the liver to the lungs
of cattle, to produce lesions of similar character. In sheep, this parasite wanders
continuously in the liver, producing black, tortuous tracts, which may be 2 cm in
diameter, and extensive parenchymal destruction. Even a few flukes may kill a sheep.
There is a single case report of natural infection in a horse.
eFigure 2-24
Destructive pigmented lesions produced by
Fascioloides magna
in an ox.
(Courtesy University of Guelph.)
The dicrocoelid flukes inhabit both biliary and pancreatic ducts. Eurytrema pancreaticum
prefers the pancreas (and is described with that organ) but in heavy infestations
can be found in the bile ducts. Dicrocoelium and Platynosomum prefer the bile ducts.
These are small, narrow flukes, 0.5-1.0 cm long, and may easily be mistaken for small
masses of inspissated bile pigment. They are not highly pathogenic, and even in heavy
infestations, there may be no signs of the toxemia observed in infestations by F.
hepatica. These flukes may occur as mixed infestations.
Platynosomum fastosum
is a small fluke inhabiting the biliary ducts and gallbladder of cats from southern
North America, Central and South America, Malaysia, and the Pacific Islands. The life
cycle includes Sublima octona snails as the first intermediate host, and Anolis spp.
lizards, marine toads (Bufo marinis), geckos, and various arthropods as second intermediate
or paratenic hosts. Cats become infected after ingestion of the second intermediate
or paratenic hosts, the cercariae are released into the upper digestive tract, enter
the biliary tree, and complete their life cycle. Mature flukes are found in the gallbladder
and bile ducts, and after 8-13 weeks, eggs are shed into the feces (Fig. 2-87
). Although the majority of infections are subclinical, heavy infestations may cause
anorexia, vomiting, lethargy, jaundice, and death. Severely affected livers are enlarged,
friable, and may be bile stained, with thickened, distended gallbladders, dilated
and/or thickened common bile ducts and cystic ducts, and dilated intrahepatic ducts.
Histologically, there are various degrees of chronic cholangitis, fibrosing cholangiohepatitis,
and cholecystitis, with dilated and hyperplastic ducts surrounded by lymphocytes,
plasma cells, and eosinophils, and in some cases neutrophils, with fluke eggs within
or adjacent to ducts. Eggs and adults may be difficult to find. Infection has been
reported to co-occur with cholangiocarcinoma in some cats.
Figure 2-87
Dicrocoelid fluke in the gallbladder of a cat.
(Courtesy Purdue University CVM.)
Dicrocoelium hospes
is found in cattle in countries south of the Sahara. Little is known of it, but it
is presumed to be comparable in all respects to the better-known D. dendriticum, which
is common in Europe and Asia and sparsely distributed in the Americas and North Africa.
Dicrocoelium spp. are found in dry lowland or mountain pastures, whereas Fasciola
spp. occur in wetter habitats, so the prevalence of Dicrocoelium is increasing with
desertification. This fluke is no more fastidious in its choice of final hosts than
many other species of fluke, and, depending on opportunity, it can infest all domestic
species, with the possible exception of cats. It is, however, of most importance as
a parasite of sheep and cattle, in which it inhabits the bile ducts. Other domestic
species and rodents are important as reservoirs.
The life cycle of
Dicrocoelium dendriticum,
the lancet fluke or small liver fluke, differs in some details from that of F. hepatica.
The eggs are embryonated when laid and do not hatch until swallowed by one of the
many genera of land snails that are the first intermediate hosts. In the snails, the
mother sporocyst produces a second generation of daughter sporocysts, which in turn
produce cercariae. The cercariae leave the snail in damp weather and are expelled
from the snail's lung, clumped together in slime balls. The slime balls are not infective
until the cercariae are swallowed by, and encyst in, the ant Formica fusca; other
ants may be involved in different countries. The cycle is completed when the definitive
hosts swallow the ants. The route of migration of the larvae from the gut to the liver
is probably via the bile ducts from the duodenum.
The pathologic changes in the liver produced by D. dendriticum are those of cholangiohepatitis
that is less severe than that produced by Fasciola hepatica (Fig. 2-88
, eFig. 2-25). The severity and diffuseness of the hepatic lesion are determined by
the number of lancet flukes present, and they may be in the thousands. The flukes
and their eggs darken the dilated ducts. Even in early infestations, there may be
some scarring of the organ at its periphery. In heavy infestations of long standing,
there is extensive biliary fibrosis, producing an organ that is indurated, scarred,
and lumpy and that at the margins may bear areas that are shrunken and completely
sclerotic. The histologic changes are the same as those in fascioliasis, with perhaps
more remarkable hyperplasia of the mucous glands of the large ducts (Fig. 2-89
).
Figure 2-88
Dicrocoelium dendriticum, the lancet fluke, in the bile ducts of a sheep.
Figure 2-89
Dicrocoelium dendriticum with embryonated eggs in the bile duct of a sheep.
eFigure 2-25
Dicrocoelium dendriticum, the lancet fluke, in the bile ducts of a sheep.
Heterobilharzia americana,
a trematode in the family Schistosomatidae, is the causative agent of canine schistosomiasis
in North America, primarily reported from the south Atlantic and Gulf Coast states.
Raccoons are the most common definitive hosts, although in addition to dogs, several
other diverse mammalian species may become naturally infected, including domestic
horses. Eggs shed in feces hatch in water and release motile miracidia, which penetrate
the tissues of the intermediate host, lymnaeid freshwater snails. Larval stages multiply
in the snail, and free-swimming cercariae are released into freshwater. These penetrate
the intact epidermis of the definitive host, migrate to the lung, and finally the
liver, where they mature, move to the mesenteric and intrahepatic portal veins to
mate, and release eggs that migrate through mesenteric venules, crossing into the
bowel lumen by means of proteolytic enzymes. Aberrant migration of eggs results in
multifocal eosinophilic and granulomatous inflammation in intestines, pancreas, and
liver. Adult trematodes may occasionally be found in hepatic vessels. Clinical signs
in affected dogs include diarrhea, vomiting, weight loss, and lethargy, and ~50% of
cases in dogs are associated with hypercalcemia, thought to be the result of unregulated
calcitriol synthesis by macrophages in the granulomatous lesions. In horses, multiple
small fibrosing granulomas scattered throughout the liver, with rare intact or fragmented
eggs, are typical and are usually clinically inapparent.
The opisthorchid flukes are parasites in the bile ducts of carnivores. They may also
occur in swine and humans, and one species, Clonorchis sinensis (Opisthorchis sinensis),
is an important human parasite. The life cycles, where known, include mollusks as
the first intermediate hosts and freshwater fish as the second.
Metorchis conjunctus is the common liver fluke of cats and dogs in North America and
is important as a parasite of sled dogs in the Canadian Northwest Territories. The
first intermediate host is the snail Amnicola limosa porosa, and the second is the
common sucker-fish Catostomus commersonii. The cercariae actively burrow into the
musculature of the fish to encyst and become infective. The immature flukes crawl
into the bile ducts from the duodenum and mature in ~28 days. Infestations may persist
for more than 5 years. Metorchis albidis has been described in a dog from Alaska,
Parametorchis complexus in cats in the United States, and Amphimerus pseudofelineus
in cats and coyotes in the United States and Panama; the life cycles are not known
but are presumed to include fish. Metorchis bilis has been reported in red foxes and
occasionally in cats from Germany.
Opisthorchis felineus (O. tenuicollis) is the lanceolate fluke of the bile ducts of
cats, dogs, and foxes in Europe and Russia. It is particularly common in eastern Europe
and Siberia and is more sparse in other areas.
Clonorchis sinensis,
the Oriental or Chinese liver fluke is an important human pathogen endemic in Japan,
Korea, southern China, and southeast Asia; dogs, cats and swine can act as reservoir
hosts. There are additional, less well-known, species of Opisthorchis in humans and
animals. The first intermediate hosts for the miracidia of O. felineus and C. sinensis
are snails of the genus Bithynia, and several genera of cyprinid fishes can act as
second intermediate hosts.
Pseudamphistomum truncatum
occurs in carnivores and humans sporadically in Europe and Asia. Its life cycle is
as for Opisthorchis.
The opisthorchid flukes, so far as known, resemble Dicrocoelium in migrating up the
bile ducts to their habitat. This may be the reason that they are more numerous in
the left than in the right lobes of the liver. They can probably live in the liver
for as long as the host lives. The pathologic effects are comparable to those of D.
dendriticum. Light infestations may be asymptomatic, and heavy infestations may cause
jaundice, chronic cholangiohepatitis, and severe biliary fibrosis. Both in humans
and animals, adenomatous and carcinomatous changes of the biliary glands have occurred
in association with these parasites; the association is probably more than coincidental.
Protozoal infections
Protozoal hepatitis is due mainly to infection with Toxoplasma (Fig. 2-90
), Neospora, and Leishmania. Granulomatous hepatitis has been described in dogs associated
with systemic infections by Hepatozoon canis. Vascular occlusion by enlarged monocytes
bearing Cytauxzoon felis merozoites can be found in the liver of infected cats. Sarcocystis
neurona schizonts were reported in the liver of a dog with systemic, multi-organ infection,
and schizonts from an unspeciated Sarcocystis were observed in hepatocytes in a horse
with suppurative and necrotizing hepatitis.
Figure 2-90
Toxoplasma gondii
cyst in the liver of a cat.
Hepatic coccidiosis causing acute cholangiohepatitis similar to that in mink and rabbits
has been observed in isolated cases in the goat, calf, and dog (Fig. 2-91
). These infections are usually considered aberrant, and often coincide with intestinal
coccidiosis. Coccidial meronts and, in some cases, gamonts are present within the
cytoplasm of biliary epithelium. The organisms are presently unclassified.
Figure 2-91
Hepatic coccidiosis in the bile duct of a pig.
Fungal infections
Fungal infections of the forestomachs with hematogenous dissemination to the liver
occur occasionally in cattle and sheep, usually as a complication of rumenitis. Lesions
in the liver are typically hemorrhagic infarcts initially or granulomatous with chronicity,
and are associated with infection by Aspergillus fumigatus and various members of
the class Zygomycetes (Fig. 2-92
).
Figure 2-92
Fungal hepatitis in a pig.
Granulomatous hepatitis associated with disseminated fungal or algal infections has
also been reported in dogs and cats. The species involved include Histoplasma capsulatum
(eFig. 2-26), Cryptococcus spp., Coccidioides immitis, Sporothrix schenckii, Aspergillus
spp., and Prototheca spp.
eFigure 2-26
Granulomatous hepatitis caused by infection with
Histoplasma capsulatum
in a dog.
(Courtesy University of Guelph.)
Further reading
Anderson PJ, et al. Resistance to Fasciola hepatica in cattle. II. Biochemical and
morphological observations. J Comp Pathol 1978;88:245-251.
Andrade RLFS, et al. Platynosomum fastosum-induced cholangiocarcinomas in cats. Vet
Parasitol 2012;190:277-280.
Arundel JH, Hamir AN. Fascioloides magna in cattle. Aust Vet J 1982;58:35-36.
Chapman BL, et al. Granulomatous hepatitis in dogs: nine cases (1987-1990). J Am Vet
Med Assoc 1993;203:680-684.
Corapi WV, et al. Multi-organ involvement of Heterobilharzia americana infection in
a dog presented for systemic mineralization. J Vet Diagn Invest 2011;23:826-831.
Corapi WV, et al. Heterobilharzia americana infection as a cause of hepatic parasitic
granulomas in a horse. J Am Vet Med Assoc 2011;239:1117-1122.
Corapi WV, et al. Natural Heterobilharzia americana infection in horses in Texas.
Vet Pathol 2012;49:552-556.
Davis CR, et al. Hepatic sarcocystosis in a horse. J Parasitol 1999;85:965-968.
Dubey JP, et al. Sarcocystis neurona schizonts-associated encephalitis, chorioretinitis,
and myositis in a two-month-old dog simulating toxoplasmosis, and presence of mature
sarcocysts in muscles. Vet Parasitol 2014;202:194-200.
Fabrick C, et al. Clinical features and outcome of Heterobilharzia americana infection
in dogs. J Vet Intern Med 2010;24:140-144.
Goto Y, et al. Frequent isolation of Echinococcus multilocularis from the livers of
racehorses slaughtered in Yamagata, Japan. Jpn J Infect Dis 2010;63:449-451.
Haney DR, et al. Severe cholestatic liver disease secondary to liver fluke (Platynosomum
concinnum) infection in three cats. J Am Anim Hosp Assoc 2006;42:234-237.
Hoon-Hanks LL, et al. Hepatic neosporosis in a dog treated for pemphigus folicaceus.
J Vet Diagn Invest 2013;25:807-810.
Jensen HE, et al. Gastrointestinal aspergillosis and zygomycosis of cattle. Vet Pathol
1994;31:28-36.
Martinez-Moreno A, et al. Liver pathology and immune response in experimental Fasciola
hepatica infections of goats. Vet Parasitol 1999;82:19-33.
McClanahan SL, et al. Natural infection of a horse with Fascioloides magna. J Vet
Diagn Invest 2005;17:382-385.
Otranto D, Traversa D. Dicrocoeliosis of ruminants: a little known fluke disease.
Trends Parasitol 2003;19:12-15.
Peregrine AS, et al. Alveolar hydatid disease (Echinococcus multilocularis) in the
liver of a Canadian dog in British Columbia, a newly endemic region. Can Vet J 2012;53:870-874.
Rezabel GB, et al. Echinococcus granulosus hydatid cysts in the livers of two horses.
J Vet Diagn Invest 1993;5:122-125.
Rushton B, Murray M. Hepatic pathology of a primary experimental infection of Fasciola
hepatica in sheep. J Comp Pathol 1977;87:459-470.
Sakui M, et al. Spontaneous Echinococcus multilocularis infection in swine in north-eastern
Hokkaido, Japan. Jpn J Parasitol 1984;33:291-294.
Schafer KA, et al. Hepatic coccidiosis associated with hepatic necrosis in a goat.
Vet Pathol 1995;32:723-727.
Taylor D, Perri SF. Experimental infection of cats with the liver fluke Platynosomum
concinnum. Am J Vet Res 1977;38:51-54.
Watson TG, Croll NA. Clinical changes caused by liver fluke Metorchis conjunctus in
cats. Vet Pathol 1981;18:778-785.
Wensvoort P, Over HJ. Cellular proliferations of bile ductules and gamma-glutamyl
transpeptidase in livers and sera of young cattle following a single infection with
Fasciola hepatica. Vet Q 1982;4:161-172.
Xavier FG, et al. Cystic liver disease related to high Platynosomum fastosum infection
in a domestic cat. J Feline Med Surg 2007;9:51-55.
Toxic Hepatic Disease
Hepatic susceptibility
The liver is particularly vulnerable to toxic injury because it is exposed to virtually
everything that is absorbed. The portal vein that drains the stomach and intestines
flows directly to the liver. This facilitates high hepatic concentrations of ingested
foreign chemicals or drugs termed xenobiotics, as well as many naturally occurring
substances with toxic potential. Most xenobiotics are unable to directly enter the
hepatocyte and require specific transporters to pass through the lipid bilayer of
the hepatocyte membrane. The uptake of exogenous and endogenous compounds from the
sinusoidal blood is facilitated by a number of basolaterally located transporters.
These transporters are members of a large group of sodium-independent solute transporters
known as organic anion transporting polypeptides (OATP). Xenobiotics may be concentrated
in hepatocytes to various degrees by mechanisms that remain obscure, but which are
associated with special binding proteins of hepatocytes, attachment to enzymatic sites
where metabolic conversions occur, and inhibition of export into the bile. The liver
is exposed to high concentrations of toxic metabolites because of its role as the
primary site of biotransformation for many therapeutic agents and endogenous substances.
Some metabolites may produce hepatocellular injury, and others may cause biliary injury
once they are transported into the canaliculi. Certain drug metabolites may be reabsorbed
in the enterohepatic circulation in a fashion similar to bile acids, facilitating
repeated exposure to the drug in question. In addition, many drug administration regimens
may achieve relatively high millimolar concentrations, resulting in depletion of conjugating
cofactors such as glutathione, which can reach the lower limits required for other
cytoprotective roles.
Role of hepatic biotransformation in hepatotoxicity
An understanding of hepatic biotransformation is essential for an appreciation of
the hepatotoxic potential of foreign compounds. Hepatocytes are the major site of
metabolism of endogenous substances and xenobiotics, including plant- or fungal-derived
secondary metabolites consumed in food, environmental chemicals and drugs. Given the
numerous enzymes and the magnitude of their expression, the liver easily exceeds the
metabolic capacity of all other organs. A major metabolic function of the liver is
to transform lipophilic substances, including endogenous steroid hormones and most
xenobiotics, into more water-soluble polar molecules to be excreted in bile or urine.
Relevant hepatic enzymes involved in biotransformation and their associated activities
can be grouped into 3 major categories.
•
Phase 1 reactions promote oxidation, reduction, hydrolysis, cyclization, and decyclization
of the parent compounds. This is typically accomplished through addition of oxygen
or removal of hydrogen, carried out by mixed-function oxidases that are usually CYP
enzymes (also known as cytochrome P450 monooxygenases), using NADPH and O2. Most of
these phase 1 enzymes are found in the smooth endoplasmic reticulum of the hepatocyte.
•
Phase 2 reactions are typically conjugation reactions in which a polar molecule, such
as glucuronic acid, sulfate, or glutathione, is added to carboxyl, hydroxyl, amino,
or sulfhydryl groups on phase 1 metabolites, making them typically less toxic and
more water soluble. Most of these enzymes are found in the cytosol.
•
In phase 3 reactions, conjugated molecules are transported by various transporter
molecules across the modified hepatocyte membrane that lines the canaliculus.
Phase 1 reactions may either increase (bioactivate) or eliminate the biological activity
of the xenobiotic substrate. However, bioactivation of molecules poses a potential
risk. Although this step is necessary to prepare the substrate to form a covalent
bond with polar compound in phase 2, it creates, if only transiently, reactive intermediates,
such as free radicals and epoxides. These reactive intermediates can bind to cellular
macromolecules, such as the CYP enzymes, that produced them, other cellular enzymes
or structural proteins, or RNA and DNA, leading to hepatocyte injury, death, or neoplastic
transformation. The CYPs can be found in all parts of the hepatic lobule, but the
hepatocytes of centrilobular region have a higher content than those of the periportal
region, which apparently accounts for the centrilobular predominance of injury produced
by compounds metabolized by this system, including carbon tetrachloride and acetaminophen.
In contrast, hepatocytes in the periportal zone are more susceptible to direct-acting
toxicants, such as metal salts, because of their proximity to incoming portal and
arterial vascular flow.
So-called “drug-drug” interactions can arise when 2 drugs are metabolized by the same
CYP, so that metabolism of one or both of the compounds is altered by competition
or interference with the relevant CYP enzyme function.
Phase 2 reactions inactivate the phase 1 metabolite through conjugation with a polar
molecule and facilitate its export by transforming the lipophilic molecule into a
water-soluble molecule that can be transported out of the hepatocyte and into the
bile or the circulation for removal via the kidney. Reduced glutathione is a major
substrate for phase 2 reactions mediated by glutathione S-transferases. Active metabolites
of many compounds, including acetaminophen are detoxified by conjugation with glutathione.
Depletion of glutathione can greatly enhance the toxicity of many compounds. Hepatic
glutathione is also important in the removal of various free radicals and reactive
oxygen species generated by normal metabolic processes as well as detoxification pathways
through the action of glutathione peroxidase. Different species and different breeds
of animals possess a divergent array of phase 1 and phase 2 types with differing levels
of activity and target substrates that contribute to the differences in metabolism
and toxicity of xenobiotics observed in different species. Acetaminophen toxicity
in the cat is a relevant example. Felids have limited phase 2 metabolism caused, in
part, from diminished activity of uridine diphosphate (UDP)-glucuronosyl transferase,
an enzyme involved in glucuronidation of bioactivated molecules. Limited glucuronidation
of acetaminophen metabolites leads to saturation of other detoxification pathways
and depletion of glutathione. The lack of glutathione then enables a highly toxic
metabolite of acetaminophen, N-acetyl-para-benzoquinoneimine (NAPQI) to bind to cellular
proteins and membranes, causing cell injury and death. NAPQI is also responsible for
the prominent methemoglobinemia seen in intoxicated cats. Toxic hepatic injury depends
on the balance between the production of reactive metabolites and their detoxification
by conjugation and other protective mechanisms.
The enzyme complex involved in bioactivation is not limited to metabolism of foreign
substances, but is also involved in the metabolism or synthesis of a number of lipophilic
endogenous substances. The principal endogenous substrates include arachidonic acid,
eicosanoids, cholesterol, bile acids, steroids, and vitamin D.
Phase 3 reactions involve movement of conjugated substrates across the membrane of
the canaliculus. As with the processes involved in uptake of substances from the plasma,
several transporters embedded in the canalicular membrane are responsible for the
movement of the water-soluble conjugated substrates from the cytoplasm into the lumen
of the canaliculus. These transporters have a range of occasionally overlapping substrates,
including bile acids and conjugates of glutathione, glucuronate, and sulfonate. These
water-soluble metabolites are excreted in the bile via members of the ATP-binding
cassette (ABC) superfamily of transport proteins located on the apical canalicular
membrane. These include the most significant transporter in humans, the multidrug-resistance
protein-1 (MDR1). Multidrug-resistance–associated protein-2 (MRP2) is responsible
for export of glutathione conjugates, such as bilirubin conjugates, and shows a striking
species difference in expression, with very low expression in the dog and very high
expression in the rat. This influences biliary excretion of drug conjugates from the
liver and may influence toxicity in the liver or elsewhere.
The mechanism by which xenobiotics and some endogenous substances can cause CYP enzyme
induction in the liver, as well as induction of phase 2 reactions, and hepatocellular
hypertrophy is mediated by the activation of nuclear receptors that function as transcription
factors. These transcription factors include aryl hydrocarbon hydroxylase receptor
(AHR), constitutive androstane receptor (CAR), pregnane X receptor (PXR), and peroxisome
proliferator–activated receptor-α (PPAR-α). These nuclear receptors are also important
mediators of hepatocellular metabolism, including hepatic lipid metabolism, bile acid
homeostasis, as well as liver regeneration, inflammation, fibrosis, cell differentiation,
and tumor formation. Each of these receptors may be directly activated by the binding
of the xenobiotic (or its metabolite) to the receptor, although some activators of
CAR do not bind to the receptor but rather phosphorylate the receptor, which results
in nuclear translocation. The nuclear receptor genes vary among species, and there
are many species differences and gene polymorphisms that affect the receptors or the
genes that they stimulate.
In summary, a broad variety of factors can influence the mechanisms and extent of
drug-induced liver injury (DILI), the term for hepatic injury from xenobiotics. There
is a variety of alternative pathways for metabolism, supported by a variety of isoforms
of different gene families, particularly the CYP enzymes and the many genetic polymorphisms
within individual alleles that code for the different isoforms. Other factors such
as age, nutritional status, sex, diet, prior or concurrent exposure to environmental
compounds, or intercurrent disease all influence the response to toxic injury. Some
of these variables contribute to the variations seen in responses among species and
individuals to liver injury.
Role of inflammation in hepatotoxicity
The extent of liver injury following exposure to xenobiotics is not, however, determined
by the chemistry of drug metabolism alone. Following exposure to injurious compounds,
the inflammatory response, particularly the innate immune response, can significantly
influence the extent of hepatocellular injury. Studies using acetaminophen have provided
most relevant information on this issue; however, the effects of inflammation are
complex, and not all studies are in agreement. There are conflicting studies demonstrating
either proinflammatory or anti-inflammatory effects of Kupffer cells following drug-induced
injury. Some clarification of this controversy has been facilitated by the identification
of subsets of intrahepatic macrophages following injury. In addition to the resident
Kupffer cells, a population of infiltrating macrophages can be detected quite soon
(within 12 hours) after acetaminophen intoxication. There are 2 major phenotypes of
intrahepatic macrophages. M1 macrophages are proinflammatory, a source for tumor necrosis
factor-α, interleukin-1β (IL-1β) and nitric oxide, which stimulates hepatocellular
injury, and these macrophages constitute the majority of the Kupffer cell population.
M2 macrophages are anti-inflammatory, secreting IL-10-, IL-6- and IL-18–binding proteins,
which modulate inflammation, and comprise the majority of the infiltrating macrophages.
Stimulation of other members of the innate immune system, such as natural killer and
natural killer T cells can accentuate acetaminophen toxicity. Neutrophils recruited
to the liver by the release of damage-associated molecular pattern (DAMP) molecules
from necrotic hepatocytes also augment tissue injury. Thus it is clear that altered
inflammation can influence drug-induced hepatic injury, but the subtleties remain
to be unraveled.
Mechanisms of injury
There are many mechanisms underlying hepatotoxicity. These include (1) covalent binding
of cellular proteins by bioactivated metabolites, which leads to intracellular dysfunction
manifested by loss of normal intracellular ionic gradients altering intracellular
calcium homeostasis, by actin filament disruption, and by cell membrane damage, such
as cell blebbing or swelling and total disruption. A consequence of actin filament
disruption can be cholestasis because of the loss of pulsatile contractions that drive
canalicular bile flow; (2) drug-induced disruption of canalicular transport pump function,
leading to cholestasis and jaundice; (3) inhibition of cellular enzyme pathways of
drug metabolism because of covalent binding of CYPs by bioactivated metabolites; (4)
covalent binding of the drug to cell proteins, which creates new adducts that serve
as immune targets for cytotoxic T-cell attack or antibody formation when transported
to the cell surface in vesicles, inciting an immunologic reaction; (5) programmed
cell death (apoptosis), occurring through tumor necrosis factor and Fas pathways;
and (6) inhibition of mitochondrial function, limiting β-oxidation of fat and ATP
generation, leading to accumulation of reactive oxygen species and lipid peroxidation,
microvesicular fat accumulation, lactic acidosis, and inability to generate ATP. These
are discussed in detail in other texts. The liver is composed of several cell types,
not only hepatocytes, and similar types of injury can also affect the biliary epithelium
and sinusoidal endothelium, and to a lesser extent, hepatic stellate cells, Kupffer
cells, and other immune cells.
Classification of hepatotoxins
Hepatotoxins can be classified as intrinsic or idiosyncratic, and these categories
apply as well to the types of drug-induced hepatotoxicity that are observed in humans
and animals. The effects of intrinsic hepatotoxins
are considered to be dose related, predictable, and reproducible in experimental animals,
and the underlying mechanisms are typically at least partially understood. Intrinsic
hepatotoxins can cause liver injury in overdose situations in most normal recipients
but are also capable of inducing similar liver damage at lower doses in individuals
with genetic or acquired abnormalities in drug metabolism. The majority of intrinsic
hepatotoxicants are converted to reactive metabolites, including lipoperoxidative
free radicals.
The analgesic acetaminophen represents a well-characterized example of an intrinsic
hepatotoxin, with species differences in metabolism and associated susceptibility
to oxidative and hepatic injury. Dogs and cats can tolerate doses within therapeutic
levels; however, higher doses may saturate the glucuronidation and sulfation detoxification
pathways, resulting in increased formation of the reactive benzoquinone-imine metabolite
NAPQI via a third cytochrome P450–mediated pathway. These can be scavenged by glutathione
conjugation; however, massive doses can cause lethal acute hepatic failure, associated
with overproduction of reactive metabolites, and depletion of hepatic and erythrocyte
glutathione. As discussed previously, clinical toxicity occurs at lower doses and
is more severe in cats because they express fewer hepatic isoforms of glucuronyltransferase
as part of phase 2 hepatic metabolism and are unable to accelerate excretion of excess
metabolites through glucuronide conjugation. This is compounded by a propensity for
hemoglobin oxidation, resulting in methemoglobinemia. Toxicity results when reduced
glutathione levels drop below a threshold level, resulting in marked oxidative stress,
and allowing reactive metabolites to bind covalently to cellular macromolecules.
By comparison, idiosyncratic hepatotoxins
are typically less dose related (although there is likely a minimum threshold), more
unpredictable, and importantly, occur in only a very small proportion of exposed individuals
(i.e., <1 in 10,000-100,000). The mechanisms of idiosyncratic hepatotoxicity are generally
not known, but reflect an unusual susceptibility of individual recipients to effects
that are unrelated to the drug's therapeutic action or overdose toxicity. Idiosyncratic
hepatotoxicity is more likely a series of rare drug-related toxicities with different
forms of pathogenesis than a single entity. There are 2 main categories of idiosyncratic
hepatotoxicity recognized in humans: hypersensitivity-related drug-induced liver injury
(drug allergy) and toxic metabolite-dependent drug-induced liver injury.
Hypersensitivity-related idiosyncrasies are characterized by a latency period before
toxicity is evident and reoccur promptly upon re-exposure. Typically, they involve
immune-mediated hypersensitivity responses to the drug metabolites that covalently
bind to liver proteins, forming neoantigens that may be recognized as foreign by the
immune system, with further liver injury resulting from the ensuing specific immune
response or upregulation of components of the innate immune system. In humans, a genetic
component to hypersensitivity-related idiosyncratic hepatic toxicity is strongly suspected.
Single nucleotide polymorphisms in the human leukocyte antigen (HLA) region or particular
HLA haplotypes have been associated with idiosyncratic drug toxicity in humans for
several drugs, reflecting the development of pharmacogenomics.
Toxic metabolite-dependent idiosyncrasies involve excessive generation of a regular
toxic metabolite, or altered metabolism to unusual hepatotoxic metabolites. There
is accumulating evidence of considerable genetic diversity (polymorphisms) in hepatic
drug metabolism among individual humans and domestic animals, and these may explain
many unusual responses to drugs. This variation could be qualitative, with the production
of a toxic metabolite not normally produced, or quantitative, with overproduction
of a normally minor hepatotoxic metabolite.
Morphology of toxic injury to the liver
The morphologic forms of hepatic injury produced by hepatotoxins are varied. Acute
toxic injury to the liver can be cytotoxic (hepatocellular), cholestatic, or mixed.
Cytotoxic hepatocellular injury results in hepatic degeneration, zonal necrosis, focal
and nonzonal necrosis (apoptosis), or lipidosis, accompanied by the clinicopathologic
features of acute hepatic injury. In general, necrosis produced by intrinsic hepatotoxins
is zonal. Acute cytotoxic injury is often manifested as steatosis (lipidosis), a result
of impairment of movement of triglycerides through the liver, interference with very-low-density
lipoprotein synthesis or transport, or impaired hepatic consumption of fatty acids
by mitochondrial oxidation. Cholestatic injury is a reflection of failure of bile
excretion associated with biliary epithelial or canalicular injury or other alteration
of bile secretion, and displays the features of obstructive jaundice. The typical
histologic manifestation consists of bile casts in canalicular spaces, with variable
parenchymal injury. An exception to this pattern can be seen in some plant intoxications,
such as those produced by steroidal sapogenins and Lantana spp., which can cause cholestasis
with minimal evidence of canalicular plugging. Mixed toxic insults display the morphologic
and clinical features of both hepatocellular and obstructive injury. Chronic toxic
injury to the liver may be manifested as chronic hepatitis, with fibrosis progressing
to cirrhosis; vascular injury, such as veno-occlusive disease; and neoplasia.
The histologic changes in acute toxic hepatic injury are rather stereotyped. They
range from apoptosis, through confluent coagulative and zonal necrosis, to massive
hemorrhagic destruction that includes sinusoidal lining cells (Fig. 2-93
). The histology of these acute intoxications is often characterized by centrilobular
necrosis, usually coagulative (Fig. 2-94
). Rarely, the pattern of necrosis may be periportal or midzonal. Depending on the
nutritional status of the animal, there may be variably severe fatty or hydropic change
in hepatocytes adjacent to the necrotic zones. The necrotic cells may accumulate calcium.
Variations on the general process of hepatocellular necrosis have little diagnostic
or pathogenetic specificity. In sublethally injured cells, there may be prominent
but nonspecific clumping of smooth endoplasmic reticulum, particularly in the centrilobular
zones.
Figure 2-93
Massive necrosis following ingestion of
Amanita phalloides
by a cat.
Figure 2-94
Acute centrilobular hepatic necrosis following ingestion of
Cycad sp. in a dog.
(Courtesy J. Cooley.)
The clinical and gross characteristics of fatal acute intoxications that destroy liver
parenchyma are rather consistent, regardless of the origin of the toxin. The animal
dies after a brief period of dullness, anorexia, colic, and various neurologic disturbances,
including convulsions; these are attributed to hepatic encephalopathy. Postmortem
examination reveals a slight excess of clear, yellow abdominal fluid, which contains
sufficient fibrinogen to form a loose, nonadherent clot. The appearance of the liver
depends on the severity and stage of the injury. Severe acute toxicity that destroys
endothelium typically results in a hemorrhagic zonal pattern, in which case the liver
may be deep red-purple and obviously swollen and turgid. In less severe injury without
hemorrhage, the liver tends to be lighter brown because of a combination of edema
(exclusion of sinusoidal blood), destruction of cytochrome pigments, and accumulation
of bile pigments and/or fat. If the animal survives for several days, the liver develops
a typical zonal yellow fatty change as triglyceride accumulates in sublethally injured
hepatocytes.
In acute fatal hepatotoxicities, there may be widespread hemorrhage resulting from
lack of coagulation factors. Petechiae and ecchymoses are seen most consistently on
serous membranes, especially on the epicardium and endocardium and abdominal viscera.
Diffuse hemorrhage into the gut, particularly the duodenum in ruminants, is also common,
as are hemorrhages into the wall of the gallbladder. Hemorrhages are largely the result
of excessive consumption of clotting factors and platelets within the areas of necrosis
in the liver, although the concurrent failure of the damaged liver to replace those
coagulation factors undoubtedly becomes important when these factors are consumed.
Gross lesions, such as icterus, and photosensitization, which reflect failure of biotransformation
or excretion of endogenous materials, develop too slowly to be a feature of acutely
fatal hepatotoxicities. However, the rate of accumulation of bilirubin increases when
hemorrhage occurs in the necrotic liver or as a consequence of coagulopathy.
Chronic hepatotoxic injury
may manifest in many patterns, as previously described. These include areas of necrosis
with primarily mononuclear inflammatory infiltrates, steatosis, cirrhosis, atrophy
with nodules, hepatic vein thrombosis, veno-occlusive disease, peliosis hepatis, cholangitis,
ductular reaction (biliary hyperplasia), and carcinogenesis. In contrast to the acute
intoxications, chronic hepatotoxicities are more likely to display a mix of these
responses, and this can provide more diagnostic specificity. For example, agents that
impair hepatocellular regeneration might not be potent necrogens but tend to produce
hepatic apoptosis and atrophy, fibrosis of various patterns, compensatory bile duct
hyperplasia, nodular regeneration, some degree of cholestasis, and frequently megalocytosis
(polyploidy). Such hepatotoxins are potential carcinogens because they favor the selective
growth of hepatocellular nodules that are resistant to mitoinhibitory effects.
Clinical signs of chronic hepatotoxicity are usually problems resulting from inadequate
detoxification and excretion; these include jaundice, photosensitization, and hepatic
encephalopathy. Most of the toxins responsible for chronic hepatotoxicity may produce
acute nonspecific zonal or massive necrosis if experimentally administered at dose
rates higher than those to which animals are likely to be exposed in the field, although
such acute toxicity is only occasionally observed in field cases of the diseases.
Toxic agents
The range of substances that can cause hepatotoxicity is so broad that it encompasses
virtually all categories of natural and synthetic chemicals. These include metals
(iron, copper), drugs (e.g., acetaminophen), plant components (phytotoxins), fungal
metabolites (mycotoxins), bacterial products (e.g., cyanobacterial microcystin-LR),
and various industrial products (especially aromatic solvents). Many drugs are also
hepatotoxic. Differences in individual susceptibility to hepatotoxic responses likely
occur for all classes of chemicals that are metabolized in the liver.
In the following sections, toxic hepatic disease will be separated into drug- or pharmaceutical-induced
hepatotoxicity (so-called adverse drug reactions), and hepatotoxicity associated with
exposure to plant or environmental toxins, including metals. The latter have been
somewhat arbitrarily divided into acute and chronic, and although it is recognized
that the difference between acute and chronic hepatotoxicity is often simply a matter
of dose rate, it is convenient to categorize the sources according to the syndrome
of liver damage that they most commonly produce. Various plants and moldy feeds are
hepatotoxic, and some phytotoxins (e.g., pyrrolizidine alkaloids) and mycotoxins (e.g.,
aflatoxins) are notorious hepatotoxins and hepatocarcinogens. Many phytotoxins and
mycotoxins target other organ systems or have physiologic rather than pathologic effects.
Some of those that cause diagnostic lesions are discussed under the respective target
tissues elsewhere. Generally, these areas of toxicology are better accessed in comprehensive
references on phytotoxins or mycotoxins.
Adverse drug reactions: drug-induced liver injury (DILI)
The definition of an adverse drug reaction is any injurious or unintended response
to a drug that occurs at a normal dose for normal use. DILI is the most common form
of adverse drug reaction in humans, and although hepatotoxic drug reactions are recognized
in dogs and cats, the true prevalence of DILI in domestic animals is unknown. DILI
should not be considered as a single disease, given the diverse array of drugs that
can trigger injury.
Acute hepatic disease has been attributed to adverse reactions to a wide variety of
therapeutic drugs, particularly in companion animals. Submassive to massive hepatic
necrosis and cholestatic hepatitis have been reported in dogs as an idiosyncratic
reaction to trimethoprim-sulfonamide combination therapy and the related sulfonamide-based
anticonvulsant drug zonisamide, although zonisamide is in a different chemical category,
is less likely to form toxic adducts, and is not likely to share a similar pathogenesis
of liver injury. Severe lobular to massive hepatic necrosis may occur in cats associated
with repeated oral administration of diazepam at recommended doses. Severe centrilobular
hepatic necrosis has been associated with use of the anthelmintic mebendazole in dogs.
Hepatotoxicity has been reported in 4 dogs treated with amiodarone, a class III antiarrhythmic
agent. This is one of the more commonly reported adverse effects of amiodarone in
humans and is related to the drug's effect on lipid metabolism. Administration of
the anabolic steroid stanozolol has also been associated with elevated alanine aminotransferase,
coagulopathy, and development of hepatic lipidosis with cholestasis in cats. Adult
beef cattle testing positive for stanozolol in urine also had hepatic changes, including
cholestasis, periportal fibrosis and inflammation, and focal necrosis, although the
changes could not conclusively be attributed to anabolic steroid administration. Other
pharmacologic agents associated with acute hepatic injury include thiacetarsemide,
methoxyflurane, halothane, oil of pennyroyal, intravenous injection of manganese chloride
and inadvertent subcutaneous injection of intranasal Bordetella bronchiseptica/canine
parainfluenza vaccine in dogs, methimazole, glipizide, and the photodynamic therapy
agent aluminum phthalocyanine tetrasulfonate in cats.
Xylitol is an artificial sweetener used commonly in baked goods or chewing gum intended
for use by diabetics or dieters. Although high doses can be tolerated by most species,
there is a particular sensitivity in dogs. Dogs that ingest >0.5 g/kg can be at risk
to develop acute severe hepatic necrosis. Dogs exposed to fatal doses have centrilobular
to massive lytic necrosis of hepatocytes and widespread hemorrhage. Affected dogs
also develop hypoglycemia and hyperinsulinemia. There may be an element of idiosyncratic
toxicity involved as dogs have tolerated higher doses in experimental settings, so
there may be breed-related or other factors influencing acute toxicity. The mechanism
of toxicity is not currently known, but intracellular ATP depletion and generation
of reactive oxygen species have been suggested as possible mechanisms for hepatocyte
injury.
Carprofen is a nonsteroidal anti-inflammatory drug used in the treatment of degenerative
joint disease and management of acute pain in dogs primarily. Diffuse and massive
hepatocellular injury, characterized by hepatocellular vacuolar change, lytic necrosis,
apoptosis, and bridging necrosis, with mild secondary inflammation and cholestasis,
has been reported in dogs administered therapeutic dosages of carprofen. The injurious
response is likely to be idiosyncratic as a chronic experimental study did not produce
injury. A related nonsteroidal anti-inflammatory drug, diclofenac, causes idiosyncratic
liver injury in humans.
The anticonvulsant drugs primidone, phenytoin, and phenobarbital have been associated
with the development of chronic hepatic disease and cirrhosis in dogs. These drugs,
used either alone or in combination, can cause biochemical and clinical signs of hepatic
dysfunction in up to 14% of dogs treated for >6 months, but only a small percentage
of cases progress to cirrhosis and hepatic failure. Currently, monitoring blood levels
and adjusting dosages of these drugs to a nontoxic level has significantly reduced
the incidence of liver injury. The most consistent histologic finding in the majority
of dogs treated with phenobarbital is proliferation of the hepatocellular smooth endoplasmic
reticulum resulting from induction of microsomal enzymes, including various subfamilies
of cytochrome P450. This results in hepatocellular swelling with fine diffuse granularity,
a so-called “ground-glass” appearance to hepatocyte cytoplasm. This histologic change
is an adaptive response, reflected clinically by increases in serum concentrations
of alkaline phosphatase, alanine aminotransferase, and γ-glutamyl transpeptidase,
but is not indicative of hepatocellular injury. Actual hepatotoxicity may represent
an idiosyncratic reaction in a small percentage of treated dogs, although the possibility
of dose-dependent intrinsic hepatotoxicity with long-term treatment has not been ruled
out. It is also possible that the enzyme induction associated with anticonvulsant
therapy may alter the ability of the liver to detoxify other nonspecified compounds
that could be the effectors of liver damage. Certainly, enzyme induction may alter
the pharmacokinetics of other co-administered drugs. Chronic hepatitis associated
with anticonvulsant therapy is characterized by bridging portal fibrosis, biliary
hyperplasia, nodular regeneration, and mild inflammatory cell infiltrates. A separate
syndrome of cholestatic hepatotoxic injury with jaundice has also been described in
dogs receiving high doses of phenytoin in combination with primidone or phenobarbital.
This is characterized by intrahepatic cholestasis, with hepatocellular swelling, vacuolation,
and small multifocal areas of hepatocellular necrosis, and has been suggested to represent
a metabolic disturbance rather than direct cytotoxic hepatocellular injury.
Hepatocutaneous syndrome
(superficial necrolytic dermatitis) associated with typical hepatic pathology of parenchymal
collapse, vacuolation, and nodular regeneration has also been reported as a separate
syndrome in dogs with a history of chronic phenobarbital therapy.
Acute and chronic hepatic disease, characterized by periportal hepatitis, periportal
fibrosis, and biliary hyperplasia, have been reported in dogs treated with oxibendazole-diethylcarbamazine
combination therapy for prevention of hookworm and heartworm. Chronic hepatic disease
has also been reported following administration of mibolerone, methotrexate and CCNU
(1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea) in dogs, ketoconazole in dogs and cats,
and megestrol acetate and griseofulvin in cats.
Hepatotoxic plants
Hepatotoxic plants occur in botanical families as diverse as the relatively primitive
Cycadaceae through the Compositae and Solanaceae. The evolutionary relationships between
herbivores and toxic plants are complex; in some situations, consumption of plants
by herbivores has competitive or neutral advantage for the plant, especially those
that are lush and prolific, so toxicity is counterproductive or unnecessary. However,
in arid or semiarid habitats, plants must put up more resistance to herbivores that
could obliterate them. Indigenous herbivores are typically reluctant to do this, and
are either resistant or reluctant to graze some plants. Plants sometimes protect critical
parts at particular stages of growth. For example, Xanthium pungens (Noogoora burr)
concentrates its toxin in the cotyledons. The seeds are rarely eaten, even by cattle,
but intoxications can occur when this plant is eaten shortly after germination. Phytotoxic
liver disease is therefore more frequently encountered in animals grazing pastures
at particular times of the year, with limited choice or supply, or when hungry animals
are introduced to plants for which their natural or induced resistance is low. The
patterns of liver disease resulting from consumption of toxic plants are for the most
part quite consistent in comparison with what is seen with exposure to other classes
of hepatotoxins. An exhaustive review of the toxicity of all such plants would be
repetitive. However, some plants that do produce more distinctive patterns of liver
lesions will be discussed in more detail.
Further reading
Adams DH, et al. Mechanisms of immune-mediated liver injury. Toxicol Sci 2010;115:307-321.
Bunch SE. Hepatotoxicity associated with pharmacologic agents in dogs and cats. Vet
Clin North Am Small Anim Pract 1993;23:659-670.
Center SA, et al. Fulminant hepatic failure associated with oral administration of
diazepam in 11 cats. J Am Vet Med Assoc 1996;209:618-625.
Court MH, Greenblatt DJ. Molecular genetic basis for deficient acetaminophen glucuronidation
by cats: UGT1A6 is a pseudogene, and evidence for reduced diversity of expressed hepatic
UGT1A isoforms. Pharmacogenetics 2000;10:355-369.
Dayrell-Hart B, et al. Hepatotoxicity of phenobarbital in dogs: 18 cases (1985-1989).
J Am Vet Med Assoc 1991;199:1060-1066.
Dunayer EK, Gwaltney-Brant SM. Acute hepatic failure and coagulopathy associated with
xylitol ingestion in eight dogs. J Am Vet Med Assoc 2006;229:1113-1117.
Harkin KR, et al. Hepatotoxicity of stanozolol in cats. J Am Vet Med Assoc 2000;217:681-684.
Holt MP, et al. Identification and characterization of infiltrating macrophages in
acetaminophen-induced liver injury. J Leukoc Biol 2008;84:1410-1421.
Jacobs G, et al. Hepatopathy in 4 dogs treated with amiodarone. J Vet Intern Med 2000;14:96-99.
Kaplowitz N, DeLeve LD. Drug-Induced Liver Disease. 3rd ed. New York: Academic Press;
2013.
Khan KN, et al. Toxicity of subacute intravenous manganese chloride administration
in beagle dogs. Toxicol Pathol 1997;25:344-350.
Kristal O, et al. Hepatotoxicity associated with CCNU (lomustine) chemotherapy in
dogs. J Vet Intern Med 2004;18:75-80.
Kumar V. NKT-cell subsets: Promoters and protectors in inflammatory liver disease.
J Hepatol 2013;59:618-620.
Leach MW, Peaston AE. Adverse drug reaction attributable to aluminum phthalocyanine
tetrasulphonate administration in domestic cats. Vet Pathol 1994;31:283-287.
MacPhail CM, et al. Hepatocellular toxicosis associated with administration of carprofen
in 21 dogs. J Am Vet Med Assoc 1998;212:1895-1901.
March PA, et al. Superficial necrolytic dermatitis in 11 dogs with a history of phenobarbital
administration (1995-2002). J Vet Intern Med 2004;18:65-74.
Marques PE, et al. Chemokines and mitochondrial products activate neutrophils to amplify
organ injury during mouse acute liver failure. Hepatology 2012;56:1971-1982.
Miller ML, et al. Apparent acute idiosyncratic hepatic necrosis associated with zonisamide
administration in a dog. J Vet Intern Med 2011;25:1156-1160.
Navarro VJ, Senior JR. Drug-related hepatotoxicity. N Engl J Med 2006;354:731-739.
Polzin DJ, et al. Acute hepatic necrosis associated with the administration of mebendazole
to dogs. J Am Vet Med Assoc 1981;179:1013-1016.
Raekallio MR, et al. Evaluation of adverse effects of long-term orally administered
carprofen in dogs. J Am Vet Med Assoc 2006;228:876-880.
Sudekum M, et al. Pennyroyal oil toxicosis in a dog. J Am Vet Med Assoc 1992;200:817-818.
Toshach K, et al. Hepatocellular necrosis associated with the subcutaneous injection
of an intranasal Bordetella bronchiseptica-canine parainfluenza vaccine. J Am Anim
Hosp Assoc 1997;33:126-128.
Twedt DC, et al. Association of hepatic necrosis with trimethoprim sulfonamide administration
in 4 dogs. J Vet Intern Med 1997;11:20-23.
Wagner M, et al. Nuclear receptors in liver disease. Hepatol 2011;53:1023-1034.
Acute hepatotoxicity: plant-derived and environmental toxins
Cyanobacteria (blue-green algae)
These primitive highly toxic microorganisms can flourish as a seasonal bloom on lakes
and ponds that have accumulated phosphates and nitrates as runoff from fertilized
soils. Outbreaks of poisoning are not common, but they occur in many countries and
may be responsible for heavy mortality among mammals or birds that drink from affected
bodies of water. Most well-documented cases have involved
Microcystis aeruginosa,
which may poison stock after the bloom has been piled by wind against the shores of
expanses of water. Other toxic species are included in the genera Anabaena and Aphanizomenon.
Some deaths are too sudden to be caused by liver damage and are probably the result
of the “fast-death factor” that has been found in some blooms. The syndrome is one
of collapse and prostration, with hyperesthesia that may be manifested as convulsions
and death within a few minutes; there are no specific lesions in this form of the
toxicosis.
The most well-understood cyanobacterial hepatotoxin is
microcystin-LR, a highly potent cyclic heptapeptide protein phosphatase inhibitor.
Other species of cyanobacteria, such as Nodularia spumigenia found in New Zealand
and the Baltic region, contain different, but still potent toxins such as nodularin.
Ruminants are most commonly poisoned, but poisoning has been reported in horses, sheep,
dogs, and domestic poultry. The cyanobacterial toxin is released when the microorganisms
disintegrate, which may occur spontaneously in bodies of water or after the application
of copper sulfate for algae control, or in the rumen or stomach after ingestion. Microcystin-LR
is very stable and persists in water at typical ambient conditions, with a 10-week
half-life. It is stable following boiling as well. The toxin is taken into hepatocytes
by organic anion-transporting polypeptide (OATP) membrane transporters and causes
cell damage by inhibiting cytoplasmic protein phosphatase 1 and 2A. This leads to
disorganization of hepatocyte and endothelial cytoskeletal actin filaments, and disruption
of their shape and integrity, leading to necrosis, apoptosis, and perisinusoidal hemorrhage.
The distribution of the necrosis is usually centrilobular to massive. The pattern
may vary within the individual liver and from case to case. In subacute intoxications,
the liver is severely fatty, and necrosis is limited to individual hepatocytes or
small groups, rather than being zonal. Phagolysosomes and bile pigments accumulate
in the cytoplasm, and there is slight biliary proliferation and fibrosis. Necrosis
of the renal tubules can also occur. Diagnosis can be made by detection of microcystin
in vomitus or liver.
Toxic fungi
Amanitins are potent hepatotoxins found in several mushroom genera, including Amanita,
Galerina, and Lepiota. The genus
Amanita
contains several species, including A. phalloides, A. verna, A. virosa, and A. ocreata,
which are considered extremely toxic. These mushrooms are mycorrhizal with various
species of deciduous and coniferous trees, and may be found in urban, suburban, and
rural areas. The amanitins are bicyclic octapeptides, ingestion of which is responsible
for gastroenteritis, hypoglycemia, and fulminant liver failure in humans, dogs, cats,
cattle, and other animals. They are stable compounds and persist in the acid environment
of the stomach and following cooking. Amanitin is transported from the systemic circulation
to hepatocytes by a specific OATP transporter on the hepatocyte surface. Toxicity
is accentuated by enterohepatic circulation of the toxin, causing repeated hepatic
exposure. Amanitin inhibits nuclear RNA polymerase II, thus interfering with transcription
and inhibiting protein synthesis, resulting in cell death. Dogs or cats dying after
ingestion of Amanita spp. have massive hepatocellular necrosis with focal areas of
hemorrhage (see Fig. 2-93). Surviving periportal hepatocytes have vesicular nuclei
with loss or fragmentation of nucleoli, consistent with ultrastructural reports of
chromatin condensation, dissolution of the nucleolus, and decline in nucleolar RNA
content. Necrosis of the proximal convoluted tubules often accompanies the hepatic
injury and can aid in the index of suspicion for amanitin intoxication. Because the
histologic lesions are nonspecific, and previously, it was difficult to obtain a definitive
diagnosis of amanitin intoxication, under-diagnosis was likely. Confirmatory testing
is available, and the liver, bile, and kidney are the preferred organs for testing.
Cycadales
Members of this order have been responsible for chronic hepatotoxicity and neurotoxicity
in cattle. Acute hepatotoxicity has been reported in cattle and sheep that have eaten
the seeds or young leaves of species of Cycas or Zamiaceae. Dogs are intoxicated by
eating the seeds of Cycas spp. usually raised as ornamental plants in warmer climates.
The toxin responsible is methylazoxymethanol, which is the aglycone of various nontoxic
glycosides, including cycasin and macrozamin, in these plants. The toxin is split
from the glycoside by bacterial metabolism in the gut, and its hepatotoxicity is the
result of further metabolism by hepatic CYPs; the pattern of necrosis is thus centrilobular
(see Fig. 2-94). The metabolites of the aglycone are apparently potent alkylating
agents, and the chronic liver lesions reflect this; there is megalocytosis (which
is not as persistent as that of pyrrolizidine alkaloid poisoning), nuclear hyperchromasia,
cholestasis, fatty change, and various degrees of diffuse fibrosis. There is fairly
consistent acute renal tubular injury. Chronic exposure leads to cancer development
in the liver, kidney, and intestinal tract of laboratory rodents.
Cycads also produce a neurotoxic amino acid, β-N-methylamino-L-alanine (BMAA). BMAA
is excitotoxic, activating neurotransmission mediated by glutamate receptors, which
in excess can lead to cytotoxic influx of calcium ions. Chronic cycad poisoning of
cattle causes a chronic nervous disorder characterized by a progressive proprioceptive
deficit. This is the result of axonopathy in upper spinocerebellar and lower corticospinal
tracts. The axonopathy, morphologically subtle at first, may eventually progress to
frank Wallerian degeneration.
Solanaceae
Toxic species of the genus
Cestrum
(jessamine) include Cestrum diurnum, a cause of enzootic calcinosis in cattle attributed
to active vitamin D analogs; the other known toxic species all produce similar hepatic
disease. Speciation within the genus is uncertain, partly because of hybridization;
however, the species named as hepatotoxic are C. parqui, C. laevigatum, and C. aurantiacum.
Cestrum spp. cause acute hepatotoxicity in the field in South America, southern and
central Africa, and Australia. Cattle are more frequently poisoned than other species,
but sheep and goats are susceptible, and fowl may be if they eat the fruit. The young
leaves and unripened berries are the most toxic parts of the plant. In acute hepatoxicity,
there is marked centrilobular and midzonal coagulative necrosis and hemorrhage. There
are no records of chronic liver disease caused by this plant, and photosensitization
is rarely seen. The toxin is water soluble and has been identified as an atractyloside.
Compositae/Asteraceae
Xanthium pungens
(Noogoora burr) in Australia and Xanthium strumarium (rough cocklebur) in the United
States, as well as X. cavanillesii, (the cocklebur) in Brazil and South Africa, have
been reported to be hepatotoxic while in the seedling stage because the toxins are
concentrated in the cotyledons. The burrs are also toxic, and although usually too
coarse to be grazed, they can be consumed if ground into feeds. Cattle, swine, and
sheep are susceptible, and toxicosis typically occurs following a period of feed scarcity,
after flooding or rain has allowed germination. The clinical signs and lesions are
not specific, being those described for acute hepatotoxins in general. The toxic principle
is a diterpenoid glycoside, carboxyatractyloside, although there may be other closely
related toxic glycosides in some plants. Wedelia glauca
also contains atractyloside and causes acute hepatotoxicity in cattle and sheep in
Uruguay and Argentina. The condition has been reproduced experimentally in sheep and
cattle and rats.
Atractyloside toxins inhibit exchange of ATP from the mitochondria with adenosine
diphosphate (ADP) in the cytosol, a process essential for oxidative phosphorylation.
Atractylosides inhibit the ADP/ATP carriers (AACs), including the form expressed in
the liver. Carboxyatractyloside blocks exchange by binding to a cationic functional
domain of bovine AAC1. Lack of ATP and mitochondrial pore leakage lead to apoptosis
and necrosis, with ion pump failure, lipid peroxidation, and glutathione depletion.
The extent of necrosis depends on dosage. In lethal poisoning with hepatic failure,
there is typically midzonal vacuolation and centrilobular necrosis, but it can be
panlobular.
Other plants produce similar lesions, but the toxins have not been identified. Helichrysum
blandowskianum is hepatotoxic to cattle and sheep in southern Australia and has caused
sudden deaths with centrilobular necrosis. The condition has been reproduced experimentally.
High mortality with acute centrilobular liver necrosis has been reported in cattle
grazing sprouting plants of Vernonia rubricaulis in Brazil. Similar lesions were reproduced
with 3 g/kg of sprouting plants. Various species of Asteraceae in South Africa, including
Asaemia axillaris, Athanasia trifurcata, Lasiospermum bipinnatum, Hertia pallens,
and Pteronia pallens, have been associated with field outbreaks of acute hepatotoxicity
in grazing sheep or cattle. Similar liver lesions have been reproduced experimentally.
Experimental intoxications by all of these species have, in some animals, produced
centrilobular as well as midzonal necrosis. Asaemia axillaris, Athanasia trifurcata,
and Lasiospermum bipinnatum are also associated with more chronic liver toxicity with
photosensitivity.
Ulmaceae
Trema tomentosa (T. aspera), the poison peach, has caused severe losses of cattle
in Australia. Similar disease has been reported in goats and horses ingesting Trema
micrantha in Brazil, and hepatotoxicity has been reproduced in rabbits. The syndrome
is acute, there is no photosensitization, and mildly intoxicated animals may recover
completely. The toxic principle is a glycoside, designated trematoxin. The pattern
of necrosis is consistently centrilobular and is identical in appearance to that of
Cestrum and Xanthium poisoning.
The gross and microscopic pathology of experimental poisoning by Trema, Xanthium pungens,
and Cestrum parqui has been shown to be identical in all morphologic respects in the
same group of sheep.
Myoporaceae
Hepatotoxic species of Myoporaceae so far incriminated are
Myoporum deserti, M. acuminatum, M. insulare, and M. tetrandum of Australia, and M.
laetum in New Zealand, southern Brazil, and Uruguay. The toxic oils are contained
in the leaves and branchlets, but within the species, there is variation in the chemical
characters and toxicity of the oils; not all strains of toxic species are toxic. There
is some delay between ingestion and absorption of the furanosesquiterpenoid oils,
the best known of which is ngaione, which are responsible for intoxication. Twenty-four
to 48 hours may elapse before signs of toxicity appear. Gross lesions include widespread
hemorrhage and a pale yellow liver. Histologically, the pattern is unusual, as the
injury to the hepatocytes occurs in periportal or midzonal regions accompanied by
biliary hyperplasia (Fig. 2-95A, B
). Some animals live long enough to become photosensitized; others may die much more
rapidly, with pulmonary edema. The edema appears to be a direct effect of the toxin
after its metabolism by the club cells (formerly Clara cells) of the airway.
Figure 2-95
Midzonal necrosis caused by ingestion of
Myoporum sp. in a donkey. A. Low-power view. B. Higher magnification.
(Courtesy R. Kelly.)
Livers from intoxicated sheep may show a striking, broad pattern of variable congestion
and even infarction, which is superimposed on the more regular lobular pattern of
periportal necrosis. Sections of these livers reveal acute fibrinoid necrosis of portal
vessels, which suggests that the coarser lesions may have a vascular basis.
Sawfly larvae
Sawfly larval poisoning is an acute hepatotoxicosis documented in cattle and to a
lesser extent sheep and goats in various locations, including Australia, Denmark,
and South America. Ingestion of the larval stage of the “sawfly,” 1 of 3 insect species
that are members of the order Hymenoptera and the suborder Symphyta, produce hepatic
injury. In Australia, the insect species is Lophyrotoma interrupta (Pergidae) or L.
zonalis (Pergidae); in Denmark the insect is Arge pullata (Argidae), and in South
America a third species, Perreyia flavipes (Pergidae), is incriminated. In parts of
northeastern Australia, the larvae are parasitic on the leaves of the tree Eucalyptus
melanophloia, and heavy infestations may occur. L. zonalis has been introduced to
Florida to control the spread of Melaleuca quinquenervi. In Denmark, sawfly larvae
feed on birch trees. There is a short clinical course following ingestion, and often,
affected cattle are found dead. Animals that survive typically develop icterus and
secondary photosensitization. At postmortem examination, ascites, petechiae, and ecchymoses
are evident. The liver is enlarged with an accentuated lobular pattern. Mural edema
of the gallbladder may be apparent. Sawfly larvae can be found in the rumen and, on
occasion, through to the abomasum. Histologically, there is centrilobular to massive
acute hepatic necrosis. Outbreaks tend to be seasonal, related to the life cycle of
the insects. The toxic principle is believed to be D-amino acid–containing peptides.
The major toxin is an octapeptide, lophyrotomin, present in the in the larvae of Australian
and Danish sawflies. There is a different toxin in the South American sawflies, identified
as the heptadecapeptide pergidin.
Halogenated hydrocarbons
Various halogenated hydrocarbons, such as carbon tetrachloride (used historically
as a fasciolicide), bromobenzene, hexachloroethane, tetrachloroethylene, and chloroform,
are activated by cytochromes P450 to hepatotoxic or nephrotoxic metabolites. They
have similar hepatotoxic properties, but these agents are little used now, so few
animals encounter them in toxic doses. However, some of these chemicals have been
used experimentally to investigate the mechanisms of hepatotoxicity, so they warrant
brief consideration here. Carbon tetrachloride is metabolized to trihalomethane, a
necrogenic free-radical metabolite, and reactive oxygen radicals are concurrently
generated. Centrilobular hepatic necrosis with midzonal hydropic change caused by
membrane peroxidation occurs within 30 hours of experimental exposures, but lethal
toxicity occurs later, when steatosis and the early stages of tissue repair responses
are manifest.
Phosphorus
White phosphorus has historically been used for vermin control, and has been implicated
in the accidental exposure and deaths of wild waterfowl. It may be present in some
incendiary devices such as fireworks. As a rodenticide, it is mixed with fat to promote
absorption, and much of the dose is transported to the liver shortly after ingestion.
A small amount may be lost by vomition, as elemental phosphorus is directly irritating
to the gastrointestinal tract. The mechanism of phosphorus hepatotoxicity is uncertain.
It is apparent that metabolism to a toxic intermediate is not necessary, but there
is some dispute on the involvement of lipoperoxidation in the hepatocellular injury.
There is evidence that protein synthesis is impaired early and that this is responsible
for the lipid accumulation that is a prominent feature. A few hours after ingestion
of phosphorus, there is severe colic and vomition. If the animal survives this acute
phase, there may be apparent recovery for a few days, followed by jaundice and other
signs of liver failure, and death by about the fifth day. At postmortem, there is
severe icterus and fatty liver, the latter sometimes being predominantly periportal
in distribution. Hepatocellular necrosis is not often a prominent feature histologically,
notwithstanding the evidence of liver failure. Fatty change is also seen in the myocardium
and distal nephrons.
Iron
Iron-dextran complexes have been widely used in the prevention and treatment of anemia
in suckling swine. Very occasionally, severe losses may occur in animals with marginal
vitamin E–selenium deficiency; in these cases, there is, apparently, iron-catalyzed
lipoperoxidation in hepatocytes and muscle. The result is sudden massive hepatic necrosis
similar in many respects to that of hepatosis dietetica. Large amounts of potassium
escape into the circulation from the liver and muscle, and sudden death may result
from the cardiotoxicity of this ion. At postmortem, there is staining of subcutaneous
tissues and lymph nodes near the site of injection, and there are lesions in the liver
or skeletal muscles. The liver is of normal size and of normal color or pale, depending
on whether or not the animal is anemic. The presence of underlying necrosis may be
indicated only by the numerous small or large hemorrhages present on the capsular
and cut surface. Insoluble iron compounds with the staining reactions of hemosiderin
are found in mesenchymal cells in many tissues, the largest amounts being in macrophages
of the local lymph nodes and in Kupffer cells. Death in piglets from hepatic necrosis
occurs ~10 hours after administration. Saccharated iron may produce acute widespread
muscle necrosis at ~24 hours, rather than hepatic necrosis in piglets with marginal
vitamin E–selenium status. The myocardium is not affected.
Acute hepatotoxicity was reported in young foals because of administration of a proprietary
paste of iron and yeast products, given as a dietary supplement within a few hours
of birth. Not all foals so treated became sick, but those that did developed severe
acute centrilobular hepatocellular necrosis, resembling the disease in piglets, and
dramatic ductular reaction (biliary hyperplasia).
Acute iron intoxication has also been reported in young cattle administered injectable
hematinics containing elemental iron, and rarely in adult horses administered oral
vitamin supplements containing ferrous fumarate or ferrous sulfate. Oversupplementation,
low vitamin E or selenium concentrations, or concurrent disease may have contributed
to these cases. Consumption of elemental iron produces a periportal or panlobular
pattern of necrosis.
Chronic hepatotoxicity: plant-derived and environmental toxins
Aflatoxin
The aflatoxins are a group of bisfuranocoumarin compounds produced as metabolites
mainly by Aspergillus flavus, A. parasiticus, and Penicillium puberulum. The metabolites
are designated by the blue or green color they fluoresce when viewed under ultraviolet
light and migration patterns during chromatography, and the major ones are B1, B2,
G1, and G2. A less toxic metabolite, M1 is found in milk and other dairy products
from cattle that ingest B1. Many others may be produced in minor amounts in fungal
colonies or as metabolic products of the major toxins in animals. The most significant
and best studied of the aflatoxins is B1
because of its relative abundance and its potency as a hepatotoxin.
Strains of
Aspergillus
differ in the varieties and amounts of individual toxins produced, indicating that
the biosynthesis of the toxins is genetically determined. The production of toxins
also varies under different conditions of fungal growth, which is influenced by the
quality of the substrate, temperature, relative humidity, moisture content of the
substrate, and microbial competition. Thus the toxicity of moldy feedstuffs is impossible
to assess without measurement of toxin production. Aflatoxins can be produced on growing
crops in the field, but much greater levels are likely to accumulate in stored or
unharvested mature grains, particularly if they are damaged by moisture. Various feeds
other than grains, ranging from legume stubbles to bread, may be the substrate in
outbreaks of aflatoxicosis. Contaminated grain has been incorporated into commercial
dog food, leading to outbreaks of acute toxicity.
Aflatoxins are metabolized by the hepatic mixed-function oxidase system to various
toxic and nontoxic metabolites, the proportions of which vary with the species and
age of the animal involved. The most potent of these is the 8,9-epoxide metabolite
of aflatoxin B1; this binds to a variety of cellular proteins causing acute toxicity,
and its carcinogenic activity derives primarily from adduct formation with guanine
in nucleic acids in sensitive species that lack adequate glutathione S–transferase–mediated
resistance. The mutational consequence is a G to T transversion. Acute toxicity is
most evident in dogs, rats, ducks, guinea pigs, and calves, and each species may be
fatally intoxicated by a dose rate of <1.0 mg/kg body weight. Although cats are also
sensitive, they rarely ingest contaminated food. Acute, fulminating liver necrosis
is sometimes seen in dogs that eat contaminated bread, dog food, or garbage, which
may contain very high concentrations of the toxin. Younger animals of all species
are much more susceptible and may die within a few hours. The gross postmortem picture
is dominated by widespread hemorrhage and massive hepatic necrosis, changes also seen
at the microscopic level (eFig. 2-27). Large animal species rarely are exposed at
sufficient doses by normal dietary intake to develop acute toxicity. Sheep and adult
cattle are quite resistant to the toxin.
eFigure 2-27
Acute aflatoxin injury in the liver of a dog, characterized by acute necrosis and
hepatic lipidosis in surrounding hepatocytes.
Prolonged exposure to low concentrations of the toxin is a more common problem than
acute toxicity in large animal species, and may merely produce reduced growth rates
and moderate enlargement of the liver without any significant hepatic signs. The enlargement
may be partly the result of hypertrophy of hepatocellular smooth endoplasmic reticulum
and some degree of fatty change. As the level of aflatoxin in the ration increases
(in young pigs, e.g., to 1.0 mg/kg ration), the liver may show all or none of the
following changes: pallor, enlargement, bile staining, increased firmness because
of fibrogenesis, and fine nodular regenerative hyperplasia. There may also be edema
of the gallbladder and bile-tinged ascites in more severe cases. Even under experimental
conditions, some individuals may show minimal liver lesions, whereas others, under
the same levels of exposure, die of liver failure. Histologically, affected livers
show obvious increase in size of some hepatocytes and their nuclei (megalocytosis)
with focal necrosis or apoptosis. Bile ductules proliferate early, and reticulin and
collagen deposition occurs throughout the acinus according to no distinct pattern
(Fig. 2-96
). Fatty change in affected livers is variable in extent and occurrence, and bile
pigments accumulate in canaliculi and hepatocytes in more severely affected livers.
Minor degrees of megalocytosis may be seen in proximal tubular epithelium in the kidney.
The changes produced resemble those of pyrrolizidine alkaloid toxicosis. This can
be attributed to the fact that aflatoxin and pyrrolizidine alkaloids inhibit hepatocellular
regeneration such that nodules and ductules regenerate as the liver becomes atrophic.
These are also genotoxic and carcinogenic, so they might be involved in the occurrence
of liver cancers in animals, as they are in humans.
Figure 2-96
Hepatocellular steatosis and ductular proliferation in canine chronic aflatoxicosis.
At higher dose rates of aflatoxin, most centrilobular hepatocytes disappear and are
replaced by a mixture of inflammatory cells, fibroblasts, and primitive vascular channels.
The liver may be much smaller than normal, particularly in young animals, presumably
because of mitotic inhibition, and focal hepatocellular necrosis is more obvious or
may be supplanted by zonal (centrilobular) necrosis. Fatty change in these livers
may be severe and uniformly distributed.
Fumonisin
Fumonisin B1, a mycotoxin elaborated by certain strains of
Fusarium verticillioides
(previously moniliforme) and
F. proliferatum
in infected corn, induces a pulmonary edema syndrome in pigs, hepatocellular carcinomas
in laboratory rats, and leukoencephalomalacia. Hepatotoxicity occurs in horses and
swine. Hepatic toxicity has also been reported in sheep and baboons. In horses, hepatoxicity
occurs less often than leukoencephalomalacia. The clinical course is relatively short,
with death likely within 5-10 days of the onset of clinical signs, such as anorexia,
depression, icterus, and edema of the head. Bilirubin and liver enzymes are typically
elevated. At postmortem, the liver is typically firm, yellow, and small, with an accentuated
lobular pattern. Histologic lesions include abundant apoptosis that may progress to
centrilobular necrosis and variable amounts of fibrosis in the portal tracts. Cardiotoxicity
is also evident in swine and horses.
Fumonisin B1 is also a primary hepatotoxin in pigs. Experimental feeding trials with
pigs have shown dose-related differences in pathology. Pigs intubated with a minimum
of 16 mg fumonisin B1/kg body weight per day developed interlobular edema, variable
hydrothorax, and pulmonary edema, whereas pigs intubated with 8 mg/kg per day for
7-8 days, or fed diets containing 200 mg fumonisin B1/kg of feed for 21 days, developed
marked icterus. Histologic changes included hepatocellular necrosis without a zonal
distribution, depletion of centrilobular hepatocytes, lobular disarray, and megalocytosis
characterized by large numbers of swollen hepatocytes with abundant granular eosinophilic
cytoplasm and occasional large nuclei, randomly interspersed with small angular hepatocytes
and scattered necrotic cells. These pigs showed no evidence of pathologic changes
in the lungs, whereas pigs given higher doses showed hepatic necrosis in addition
to pulmonary edema. The liver changes appear to be reversible upon cessation of exposure.
Gilts fed low levels of fumonisin B1 for 90 days developed hepatic nodular hyperplasia,
along with hyperkeratosis and parakeratosis and hyperplasia of the distal esophageal
mucosa. Fumonisins are inhibitors of sphingosine and ceramide synthetase, leading
to inhibition of sphingolipid biosynthesis. As a consequence, there is an accumulation
of bioactive intermediates of sphingolipid metabolism (sphinganine and other sphingoid
bases and derivatives), as well as the depletion of complex sphingolipids, which interfere
with the function of some membrane proteins and may be involved in the toxic hepatic
effects.
Phomopsin
There are 2 distinct manifestations of toxicity associated with Lupinus spp.; discussed
here is the condition formerly known as “lupinosis,” which is a true mycotoxic liver
disease. The teratogenic and neurotoxic effects of some of the isoquinoline alkaloids
from the plants themselves are discussed in Vol. 1, Bones and joints and Vol. 1, Nervous
system.
The fungus
Diaporthe toxica (formerly Phomopsis leptostromiformis) is parasitic on green Lupinus
plants, but it becomes saprophytic after the host plant dies. Phomopsins (A or B)
are produced if the lupin stubble is moistened, and such stubbles may remain toxic
for months. Additional toxins are suspected. Severe acute liver damage has been described
in sheep on very toxic stubbles in Western Australia, but in many of these cases,
it has been difficult to separate the toxicity of the lupins from that of copper,
which in this area is often concentrated in ovine livers (see later section on Copper).
The usual syndrome of phomopsin poisoning is subacute to chronic. Inappetance occurs
soon after experimental administration of the toxin is begun; liver damage is clinically
inapparent for several days. The gross pathologic abnormalities of acutely intoxicated
sheep include icterus and modest ascites. The main abnormalities are evident in the
liver, which varies from a yellow discoloration because of lipidosis in more acute
cases, to a variable ochre to orange discoloration with a firm texture with increasing
chronicity (Fig. 2-97A
). Histologically, there is early hepatocyte swelling and accelerated cell death among
hepatocytes. Increased mitotic activity is soon apparent, although by this time, the
liver has become smaller. The mitotic activity is in fact largely ineffectual, as
close examination reveals that many mitotic figures are abnormal. There is either
clumping or dispersal of chromatin, and there appears to be mitotic arrest at late
metaphase (Fig. 2-97B- D). Remaining hepatocytes swell, their cytoplasm becomes granular,
and their nuclei vesicular and may contain vesicular intranuclear pseudoinclusions.
There is variably severe fatty change, dependent to a large degree on the fat reserves
of the animal. There is also accumulation of complex pigment in macrophages in portal
stroma and about hepatic venules; this granular material contains lipofuscin, ferric
iron, and copper at least. Bile duct proliferation is also a prominent feature of
the chronic disease. With progression, hepatic fibrosis occurs, predominantly diffuse
in distribution, and by this stage, there is usually clinical icterus and anorexia.
Figure 2-97
Phomopsin poisoning (lupinosis) in sheep. A. Affected sheep have pale livers with
prominent icterus. B. Numerous, and abnormal, mitotic figures in subacute phomopsin
poisoning. C. Brown pigment within Kupffer cells. D. Clear intranuclear pseudoinclusions
can be found in affected hepatocytes.
(Courtesy J.G. Allen.)
The liver continues to shrink, presumably as a result of continued mitotic inhibition
and progressive fibrosis. The organ is small, tough, and has a finely granular surface
and texture. It is pale gray-orange but usually retains its shape. Fibrosis is initially
portal to periportal, but central areas can also become fibrotic. Bridging fibrosis
between portal areas and portal to central regions can develop. In naturally occurring
cases, however, discontinuous intake of the toxin may produce a liver grossly distorted
by asymmetrical nodular regeneration and fibrosis. The atrophic changes are most severe
in the left lobe.
Photosensitization occurs in phomopsin-poisoned sheep; it may be severe if the animals
have access to green feed while under the influence of the toxin. Lupinosis in sheep
has also been experimentally observed to induce mild skeletal myopathy, resembling
nutritional myopathy.
Phomopsin poisoning in cattle causes most losses when the animals are lactating or
heavily pregnant; in these animals, the syndrome is essentially one of ketosis, to
which such cows would be predisposed by the anorexia that is an obvious clinical feature
of this intoxication. Pregnant sheep are less likely to have access to toxic lupin
roughage during late gestation; otherwise, ketosis triggered by phomopsin would be
expected just as often as in cattle.
Chronic hepatic fibrosis with fine, nodular regeneration may occur infrequently in
cattle as the result of chronic phomopsin poisoning. Similar liver changes may also
be seen in horses, in which there may also be hemolytic anemia of unknown pathogenesis.
Sporidesmin
The mycotoxin sporidesmin is produced by the fungus
Pithomyces chartarum
and concentrated in the conidia (spores)
;
the most important substrate is dead ryegrass (Lolium perenne) that has been moistened
in warm weather. Intoxication causes chronic liver damage and severe hepatogenous
photosensitivity (“facial eczema”)
and is a serious cause of loss of sheep and, to a lesser extent, cattle, goats, and
farmed deer on the North Island of New Zealand. Sporadic and subclinical intoxication
occurs irregularly on the South Island, in southern Australia, and South Africa. Although
the fungus is found throughout the world, toxigenic strains appear to be limited to
New Zealand. Sporidesmin intake in conjunction with ingestion of Tribulus terrestris
causes another hepatogenous photosensitivity (geeldikkop); this is similar to but
distinct from facial eczema, and is described later.
Sporidesmin is concentrated in the fungal spores, and the toxigenicity of pasture
is related to the density of the spores in it. Sporidesmin is not specifically hepatotoxic.
Administration of the toxin does produce rapid disorganization of hepatic cell organelles
and triglyceride accumulation, but these are mild and nonspecific changes. If administered
in suitable dosage, the toxin causes permeability alterations in many tissues and
will, for example, produce corneal edema on local application. The hepatobiliary lesions
are caused by the excretion of unconjugated sporidesmin in bile, where its concentration
may initiate oxidative injury, likely mediated by the production of a hydroxyl radical.
Sporidesmin is also excreted in urine, and if the dose is high enough, edema and mucosal
hemorrhage occur in the bladder. The hepatic lesion is the result of irritation of
mesenchymal tissues in the portal triads and surrounding the bile ducts. A high concentration
of sporidesmin can injure biliary epithelium, allowing diffusion of the bile duct
contents. Release of toxin, possibly accentuated by the release of bile acids as well,
produces irritative lesions and necrosis in the adjacent blood vessels.
The liver in acute forms of the disease is enlarged, with rounded edges, and is finely
mottled and discolored yellow-green by retained bile pigments, although the discoloration
may be blotchy. There is mild edema and congestion of the wall of the gallbladder,
which may be distended with bile of normal quality or with mucin (white bile). The
extrahepatic ducts are thickened and prominent, and there is edema of the adventitia.
The ductal changes may extend to the papilla of Vater and can be traced by the naked
eye deeply into the parenchyma. In more chronic cases, alterations of size and pigmentation
of the liver are variable. Pale areas of capsular thickening, which may be elevated
or depressed, are visible. On cut surfaces, they extend deeply as wedge-shaped areas
in which biliary fibrosis has produced an exaggerated acinar pattern, and the parenchyma
is pale and atrophic; these areas are related to occluded bile ducts.
The liver is firm and cuts with increased resistance. The medium and large caliber
intrahepatic ducts are conspicuous. There is irregular stenosis of their lumens, some
are occluded by cellular debris and inspissated bile or mucin, and in some, cicatrization
of the new fibrous tissue causes complete atresia. Occlusion of the ducts causes the
parenchyma served by them to undergo atrophy, necrosis, and fibrosis (Fig. 2-98
). The livers of animals that have survived an attack of cholangitis of this genesis
are distorted in shape and size by large nodules of regeneration and persistent areas
of atrophy and fibrosis. The atrophy and fibrosis may affect either lobe, but usually,
the left is most severely affected.
Figure 2-98
Chronic sporidesmin intoxication (“facial eczema”) in sheep. Liver lobe atrophy and
fibrosis.
(Courtesy K.G. Thompson.)
Histologically, the changes are those of acute cholangitis or cholangiohepatitis to
which there is minimal leukocytic reaction. There is extensive necrosis of the lining
of the larger intrahepatic ducts and the extrahepatic ducts, the epithelium being
cast off as debris mixed with a few leukocytes. There is edema of the adventitia of
the ducts, with active fibroplasia and scarring. Inflammatory cells are present, but
not in large numbers, and they are chiefly lymphocytes and histiocytes. Injury to
the smaller radicles of the bile ducts is less severe, but fibrosis is active. The
portal tracts are enlarged by fibrous tissue and by the active generation of new bile
ducts that follows and which is more prominent in chronic intoxication (eFig. 2-28).
Affected camelids appear to be susceptible to florid hyperplasia of the bile ducts.
eFigure 2-28
Chronic sporidesmin toxicity in sheep causes prominent ductular fibrosis of larger
ducts and damage to the biliary epithelium.
(Courtesy R. Kelly.)
In more severe intoxications, there may be coagulative necrosis of blood vessel walls
in the portal triads; when this is incomplete, the most damaged segment of the vessel
tends to be that adjacent to the nearest injured bile duct. Both arteries and veins
may be affected, and it is possible that necrosis is related to vascular insufficiency
as well as to impaired bile drainage. Changes in the hepatic parenchyma are minimal
and secondary to those in the portal triads. In acute cases, there may be extensive
pigmentation of hepatocytes and Kupffer cells by bile pigments, but this is irregular
in distribution in the liver. Inspissated bile can be found in the bile ducts and
canaliculi. There is some necrosis of hepatocytes adjacent to inflamed portal areas,
and other areas of necrosis, focal in type and distribution and probably the result
of biliary obstruction, may be numerous.
Other morbid alterations include great enlargement of the adrenals produced by cortical
hypertrophy; sclerotic intimal plaques in the arteries, veins, and lymphatics in the
hilus of the liver; and a tendency for the newly formed bile ductules to recanalize
occluded ducts.
Pyrrolizidine alkaloids
Pyrrolizidine alkaloids have been identified in nearly 3% of all plant species, from
>6,000 species of 3 families: the Asteraceae (Compositae), Leguminosae (Fabaceae),
and Boraginaceae. The main genera responsible for plant toxicoses in domestic mammals
are
Senecio, Crotalaria, Heliotropium, Cynoglossum, Amsinckia, Echium,
and
Trichodesma,
and are widely distributed around the world. Intoxication is relatively infrequent
because most plants containing pyrrolizidines are unpalatable. Contamination of baled
or cubed forage with toxic plants is a common route of exposure. The seeds are also
toxic, and the small seeds of Amsinckia and Crotalaria may, depending on harvesting
technique, heavily contaminate other harvested grains used in prepared pig and poultry
feeds. More than 350 pyrrolizidine alkaloids have been identified chemically, and
most of the toxic plant species contain more than one of the alkaloids. So far, only
a small proportion of the known alkaloids have well-characterized toxicity; these
are all esters of 1 of 3 amino alcohol bases (necines) or acids (necic acids). The
retronecine group includes monocrotaline, retrorsine, retronecine, ridelliine, senecionine,
and jacobine, and the heliotridine group includes lasiocarpine and heliotrine. These
toxins must be metabolized to more reactive forms for toxicity. Cytochromes P450 mediate
the N-oxidation of the necine bases. They can also mediate the 2-step hydroxylation
of the necine bases at the C3 and C8 positions; this is followed by spontaneous dehydration
to the highly reactive dehydropyrrolizidine (DHP) alkaloids. The toxic DHP alkaloids
are electrophilic and can bind covalently to amino acids, proteins, and nucleic acids
at guanine and adenine residues. Acute toxicity derives from damage to cellular proteins.
Antimitotic effects can be exerted via damage to microtubules. The DNA-binding activity
is responsible for genotoxicity; the DHP-derived DNA adducts are a common pathway
for carcinogenic activity of pyrrolizidine alkaloids. The DHP metabolites can be detoxified
by glutathione conjugation by glutathione S–transferases. Ester linkages at the C7
and C9 positions can be hydrolyzed by carboxylesterases to generate the necine or
acid moieties; this is generally considered to be a detoxification pathway.
The toxicity of a pyrrolizidine alkaloid-containing plant depends on many factors.
The content of toxin varies by species of plant; the stage of growth, as new growth
tends to have more toxin in general; and the time of year and environmental factors
such as rainfall. For an individual pyrrolizidine alkaloid, toxicity depends on the
amount of alkaloid that can be converted to reactive metabolites, the rate of cytochrome
P450–mediated generation of the DHP, and the efficiency of detoxification by glutathione
conjugation. Toxicity also depends on the species exposed, age and sex of the animal
intoxicated, and the metabolic activity and the mitotic stage of its target cells.
Young animals are generally much more susceptible than adults. Grazing animals are
more likely to consume plants that contain pyrrolizidine alkaloids, but because the
toxins are produced by the plants to deter herbivores, it is unusual for an animal
to consume large amounts. Ruminants, especially sheep and goats, are much less susceptible
than pigs, in part because the toxins can be degraded in the rumen. However, sheep
can graze out stands of Senecio that would be lethal to cattle. Horses and cattle
have similar, intermediate susceptibilities. Most pyrrolizidine alkaloids are hepatotoxic
because they are metabolically activated to DHPs in the liver. There are 3 common
morphologic expressions of pyrrolizidine poisoning. First, acute centrilobular to
massive necrosis occurs in animals ingesting large amounts of these alkaloids. Because
the plants responsible are unpalatable, naturally occurring outbreaks of acute poisoning
are generally restricted to circumstances where animals face starvation, such as grazing
pastures affected by prolonged drought. Ingested dosages in grazing circumstances
are usually too low for acute effects; however, acute hemorrhagic centrilobular necrosis
has been described after experimental exposures to high doses of pyrrolizidine alkaloids.
The centrilobular pattern of necrosis produced is similar to that caused by many other
hepatotoxins that are bioactivated by hepatic cytochromes P450. Second, phasic (usually
seasonal) repetitive exposure to these alkaloids leads to hepatic atrophy with formation
of regenerative nodules. This is the most common expression of field exposure to pyrrolizidine
alkaloids, and affected livers show a characteristic pattern of hepatocellular polyploidy
known as
megalocytosis (Fig. 2-99
). Third, prolonged exposure exclusively to Heliotropium induces firm, fibrotic atrophic
livers without nodular regeneration. If seasonal exposure to these toxic alkaloids
declines, then a remarkable degree of recovery is possible.
Figure 2-99
Megalocytosis in Senecio ragwort poisoning in an ox.
Pyrrolizidine alkaloids inhibit DNA synthesis and mitosis in hepatocytes, but some
are able to replicate their DNA without undergoing mitosis, resulting in greatly enlarged
hepatocytes with large convoluted polyploid nuclei. Some enlarged nuclei have cytoplasmic
invaginations that can become entrapped as intranuclear inclusions. However, many
hepatocytes in an affected liver do not become megalocytic. Those that are completely
inhibited do not replicate DNA at all, whereas those that are more resistant can replicate
more normally and give rise to nodular populations of smaller, more normal hepatocytes.
Inhibited hepatocytes and megalocytes are long lived but eventually many undergo apoptosis.
In chronic pyrrolizidine alkaloid poisoning, the liver can become atrophic as hepatocytes
are lost faster than they can be replaced. The atrophy can be compensated to various
degrees by megalocytosis and regeneration of small, less inhibited hepatocytes that
proliferate in a nodular pattern. Concurrently, there is usually proliferation of
bile duct epithelial cells (termed ductular reaction) in the portal triads. This is
largely explained by the propensity of hepatic progenitor cells to proliferate forming
ductules when adult hepatocytes cannot respond to the regenerative stimuli that prevail
when liver mass is inadequate. There may also be some periportal fibroplasia that
varies with species and exposure; typically, it is minimal in sheep, moderate in horses,
and may be marked in cattle (eFig. 2-29). In cattle, the fibrous tissue can infiltrate
along the sinusoids to dissect lobules, separate individual cells, and link up with
the walls of efferent veins. In acute poisoning, in which there is centrilobular necrosis,
fibrosis can also develop in a “veno-occlusive” pattern around and sometimes obliterating
the hepatic venules. There is evidence that pyrrolizidine metabolite injury is not
restricted to hepatocytes and that endothelial cells are also involved, leading to
additional vascular insult and fibrosis in the liver. Direct endothelial injury from
pyrrolizidines may occur, but an alternative explanation proposes a form of “bystander”
injury, where toxic metabolites are formed in hepatocytes but are able to damage adjacent
endothelial cells. This pattern of fibrosis may occur in some field cases in cattle,
but is more commonly observed in humans exposed to pyrrolizidine alkaloids in herbal
preparations or “bush teas,” and it can be produced experimentally after sublethal
acute experimental hepatotoxicity. In fatal cases in cattle, the livers are very tough
and nodular, and the nodules have variably efficient biliary drainage and cytochrome
expression, so there may be a very striking color pattern, ranging from fatty yellow
through green and brown. Hepatic fibrosis can result in portal hypertension with ascites,
severe mesenteric edema, and diarrhea. Excretory insufficiency with moderate jaundice
and photosensitization can also occur.
eFigure 2-29
Diffuse fibrosis of liver in chronic
Senecio poisoning in an ox.
(Courtesy P. Stromberg.)
The disease in sheep is always protracted as a consequence of the relative resistance
of this species; indeed, clinical signs may not be seen until after a second season
of exposure. The plants most often implicated are Heliotropium europaeum and Echium
plantagineum. The shape of these failed livers is normal, but they are small, gray-yellow,
fairly smooth, and toughened by condensation of normal stroma rather than by fibroplasia.
If the liver copper content was high before intoxication, toxicity can culminate in
copper release and an episode of intravascular hemolysis. In this case, the carcass
will be intensely jaundiced, and the kidneys are stained with methemoglobin and bilirubin.
The relationship of chronic copper poisoning to pyrrolizidine alkaloid poisoning is
discussed later.
Horses are susceptible to both acute and chronic toxicosis, and liver failure produced
in horses by these alkaloids is similar to that seen in cattle. Horses are more likely
to manifest signs of hepatic encephalopathy with head-pressing and compulsive walking;
in some places, these nervous signs give rise to colloquial names such as “walkabout”
and “walking disease.” Administration of a high dose of Cynoglossum officinale resulted
in severe liver disease within 7 days after dosing, with elevated serum enzymes, altered
bile acid metabolism, and extensive hepatocellular necrosis with minimal periportal
fibrosis and biliary hyperplasia, and little or no megalocytosis. Edema and infarction
of the cecum and colon were also present at postmortem. Administration of a low dose
to horses for 14 days resulted in transient clinical depression and weight loss, transient
elevations of serum enzymes and bile acids, and minimal periportal hepatocellular
necrosis with fibrosis, developing extensive megalocytosis by week 14. The megalocytosis
became the most prominent change 6 months after exposure.
Metabolites of pyrrolizidine alkaloids are formed by any cells with adequate cytochrome
P450 activity. Although the liver is the main site of bioactivation, other cells such
as proximal convoluted renal tubules and club cells (formerly termed Clara cells)
in the lung can generate metabolites that cause local injury. Death in some instances
may be the result of renal damage and in others the result of pulmonary vascular and
interstitial lesions. Variation in the source of the toxin and in the species-based
metabolic differences in the affected animals account for the differences in susceptibility
of the different tissues. Alkaloids from Crotalaria affect the widest range of tissues
in most animals; most notably, monocrotaline causes diffuse lung injury, leading to
pulmonary edema progressing to fibrosis. Respiratory difficulty has been described
in horses eating C. dura, and C. crispata produces similar lesions. Sheep develop
pulmonary signs after eating C. globifera and C. dura, and pigs after eating Senecio
jacobaea. The experimental feeding of C. spectabilis to rats or the injection of monocrotaline,
extracted from the plant, produces progressive pulmonary disease, pulmonary hypertension,
and cor pulmonale, with necrotizing vasculitis of the pulmonary arterioles. Emphysema
occurs in pigs and is an outstanding feature of the pulmonary disease of horses. The
essential reactive lesion is diffuse fibrosis of alveolar and interlobular septa,
with patchy epithelialization occurring more slowly.
Lantana camara
Lantana camara is an attractive ornamental shrub native to the Americas and Africa
that grows readily in tropical and subtropical habitats. It contains various toxic
pentacyclic triterpenes, the most abundant of which are lantadene A, lantadene C,
and icterogenin, although metabolites may also be toxic. Lantadene A appears to be
the most toxic; at high doses experimentally, it causes severe acute centrilobular
necrosis of the liver. The mechanism of toxicity is not known, but may be related
to the effects of lantadene A on mitochondrial energetics. However, L. camara poisoning
in grazing animals is manifested as subacute or chronic cholestasis, characterized
by severe icterus and photosensitization, most commonly seen in cattle, but rarely
in sheep, goats, and horses. Goats are quite susceptible to the toxin, but are less
likely to eat the plant. Neonatal ruminants appear resistant suggesting the rumen
may retain the plants and provide longer exposure to the toxins.
Photosensitization is usually severe after 2 days, but jaundice is more severe in
chronic cases. Heavily intoxicated cattle can die within 2 days, but most fatal cases
run a course of ~2 weeks. Ruminal stasis and anorexia appear early, and the large
bowel contains dark, dry feces. The rumen is a reservoir of toxin that remains active
and can perpetuate the intoxication after access to the plant is curtailed. The liver
is enlarged, pale, and stained yellow, orange, or green-gray by bile pigment. The
gallbladder is greatly distended with pale, sometimes slightly mucoid, bile.
The severity of the liver changes may be much less than expected from the intensity
of the icterus and photosensitization. The most consistent histologic finding in the
liver is hepatocellular enlargement and fine cytoplasmic vacuolation, together with
some degree of bile accumulation in canaliculi, hepatocyte cytoplasm, and Kupffer
cells. The canalicular cholestasis is usually more severe in the centrilobular zones,
whereas the cytoplasmic vacuolation is often more pronounced in the periportal hepatocytes.
There is usually some bile duct proliferation, and in some cases, there will be a
high incidence of periportal apoptosis, focal coagulative necrosis, or hepatocellular
dissociation.
Electron microscopy reveals an apparent increase in volume of smooth endoplasmic reticulum
and a quite characteristic form of collapse of many bile canaliculi. Other canaliculi
are distended and have damaged microvilli. Because much of the bilirubin that accumulates
in the plasma of such animals is conjugated, it seems that the cholestasis is due
in large measure to direct interference with canalicular transport of bile. The mechanisms
of cholestasis have not been determined, but damage to the contractile pericanalicular
cytoskeleton or its associated cell adhesion molecules required for canalicular integrity
are plausible targets (see previous section on Cholestasis and jaundice).
Animals are usually polyuric because of acute tubular injury, resulting in severe
dehydration, in part related to a disinclination to drink. The kidneys are slightly
enlarged and wet on section, and, especially in the more chronic cases, the cortex
is pale and the medulla hyperemic. The renal lesion is nonspecific acute tubular injury,
ranging in severity from mild vacuolar change to patchy tubular necrosis and extensive
tubular cast formation. The role of the hyperbilirubinemia in the production of the
renal damage has not been assessed. Severe myocardial necrosis can be produced in
sheep by Lantana poisoning, and may be responsible for the early deaths in cattle.
Intoxication by steroidal sapogenins: tribulosis and related toxicoses
Consumption of
Tribulus terrestris,
particularly by the grazing of young wilted plants or sometimes hay, causes cholangitis
and photosensitization in sheep. This plant has caused enormous loss of sheep in South
Africa, where the toxicosis is known as geeldikkop (“yellow bighead,” because of icterus
and marked edema of ears and face). The disease has been reproduced by oral administration
of crude extracts of steroidal sapogenins or their glycosides, saponins, of T. terrestris,
but which of the various saponins in the plant are responsible for the disease is
unknown. The disease has also been reproduced experimentally by co-administration
of sporidesmin and T. terrestris, but the liver lesions are histologically distinct
from those produced by sporidesmin alone (facial eczema).
In geeldikkop, the most characteristic gross finding is the presence of a white, semifluid
accumulation of fine, crystalline material that can be expressed from the cystic duct
and larger intrahepatic ducts. The gallbladder mucosa is also partly covered with
a fine, crystalline deposit. The gross lesions of geeldikkop are similar to those
of facial eczema in that there is generalized icterus, and the liver is discolored
by bile pigment and either slightly swollen or distorted, according to the duration
of the disease.
In poisoning by sporidesmin alone, however, there is more obvious edema and fibrosis
of bile ducts, and bile infarcts in the parenchyma are more common. The acute lesion
consists of swelling and feathery vacuolation of hepatocytes, and marked hyperplasia
of Kupffer cells that may show similar cytoplasmic changes. In these acute cases,
the presence of acicular crystals may be very difficult to detect (Fig. 2-100A, B
). With more chronic intoxication, the most consistent histologic abnormality is the
presence in bile ducts of various amounts of crystalline material (Fig. 2-100C). The
crystals are fine, flat, and are deposited in affected ducts, and, less commonly,
in hepatocytes themselves and in renal tubules. There may be associated bile ductular
proliferation in severe cases, but often the degree of histologic hepatocellular damage
is mild compared to the severity of the photosensitization. The cholestasis is not
solely the result of mechanical obstruction by the crystals, because photosensitization
can occur in such outbreaks in animals whose livers show very little cholangitis and
contain very few crystals. The severity of peribiliary fibrosis is more variable and
probably depends on the relative contribution of sporidesmin to the intoxication.
There is some hepatocellular degeneration and apoptosis, and there is fairly uniform
swelling of cytoplasm. Bile pigment accumulates in Kupffer cells and hepatocyte cytoplasm,
but not to any marked extent in canaliculi. Focal necrosis of the gallbladder mucosa
is often present. The severity of the hepatic lesion increases with the duration of
exposure.
Figure 2-100
A. Crystal deposition within bile duct epithelium in a goat grazing
Panicum sp. B. Foamy material within macrophages in an ox ingesting
Brachiaria sp.
(Courtesy R. Kelly.)
C. Fine crystalline material in expanded Kupffer cells in a goat grazing
Panicum sp.
The biliary crystals from sheep with geeldikkop have been determined to be composed
principally of calcium salts of steroidal sapogenins present in T. terrestris. Plant
saponins are metabolized in the rumen and liver to episapogenin glucuronides, which
in the presence of calcium may precipitate, forming the characteristic biliary crystals.
Cholestasis is likely related to reduced bile acid secretion into the lumen of the
canaliculi, rather than biliary occlusion by these plant sapogenins. This explains
the evidence of cholestasis before crystals can be seen histologically. Other unidentified
plant components may also play a role in hepatocellular and biliary injury. It has
also been suggested that the toxins responsible may act primarily on the membranes
of the bile canaliculi, in a manner similar to that of Lantana poisoning. Hepatocellular
damage likely also plays a role in phytoporphyrin (phylloerythrin) retention.
Switchgrass (Panicum virgatum), Kleingrass (P. coloratum), and other
Panicum species are associated with hepatogenous photosensitization in grazing animals,
including sheep, goats, and horses in the United States, Australia, and other areas
of the world. The toxic agent(s) are steroidal sapogenins, and their type and concentration
vary with the part of the plant (highest concentrations being in the young growing
leaf) and with the environmental conditions. Hot and dry conditions, have been associated
with outbreaks. Younger animals appear to be more susceptible. Crystals may be produced
in the biliary tree, hepatocytes, sinusoids, or Kupffer cells.
Crystal-associated cholangiohepatopathy is not specific for Tribulus or Panicum spp.
intoxication. Similar changes occur in the hepatogenous photosensitivity disease,
alveld, in sheep grazing pastures in Norway, the British Isles, and in the Faeroe
Islands containing Narthecium ossifragum. Similar disease has been reported in ruminants
intoxicated by Agave lecheguilla, Nolena texana, and the pasture species Brachiaria
decumbens (signal grass) in Brazil. In New Zealand, crystal deposition in the biliary
tree of cattle has been reported following ingestion of Phytolacca octandra (inkweed).
In those cases studied (T. terrestris, P. dichotomiflorum, P. schinzii, Narthecium
ossifragum), the characteristic crystalloid material deposited in the bile is principally
composed of calcium salts of steroidal sapogenins. In some cases, crystal formation
may not be apparent in bile ducts, and affected Kupffer cells and macrophages are
more evident. There may be differences in the histologic responses of cattle and sheep
grazing various sapinogenin-containing plants. Foamy Kupffer cells and hepatocytes
along with foamy macrophages in mesenteric and hepatic lymph nodes may be a more common
response in cattle versus crystal formation within bile ducts in sheep (Fig. 2-101
). To complicate diagnostic efforts, foamy macrophages in the mesenteric and hepatic
lymph nodes are also found in healthy cattle and sheep grazing B. decumbens in Brazil.
With the exception of Nolena texana, steroidal sapogenins have been demonstrated in
all of these plants. As with T. terrestris, extracts of B. decumbens containing steroidal
saponins produced typical lesions of cholangitis with crystal deposition when orally
administered to lambs, with Pithomyces chartarum spore counts below detectable levels.
Similarly, analysis of samples of B. decumbens and P. dichotomiflorum on which cattle
and goats had recently been photosensitized showed only low levels of P. chartarum
spores, and all isolates obtained failed to produce sporidesmin. Despite this, it
is also apparent that neither Tribulus nor Panicum stands are at all times dangerous.
Levels of saponins have been shown to vary greatly within a species from site to site,
and with the age of the plant. Additionally, the role of sporadic environmental conditions,
such as wilting, which might concentrate the saponins or other unidentified toxins,
or the synergistic involvement of endophytic fungi, remains to be determined.
Figure 2-101
Chronic cholangitis and plugging of bile duct by lipophilic crystalloid material,
in photosensitivity disease of sheep grazing pangola grass. Similar to reaction seen
in geeldikkop.
(Courtesy J.G. Allen.)
Other plants reported occasionally to produce unexpected photosensitivity include
Digitaria spp., Cooperia pedunculata, Nidorella foetida, and Chloris spp., and such
valuable pasture genera as Medicago, Trifolium, Avena, and Biserrula pelecinus, although
the latter species may cause primary photosensitization.
Nitrosamines
Epizootics of poisoning by dimethylnitrosamine occurred in Norwegian cattle, sheep,
and fur-bearing animals, from 1957-1962. The toxin was present in herring meal and
was thought to be a reaction product of trimethylamine and other lower amines with
sodium nitrite, added as a preservative, the reacting amines being products of decomposition.
Animals consumed several pounds of the toxic meal each day before becoming ill. This
intoxication has little importance in livestock now that it is easily prevented.
At postmortem, there was moderate anasarca and signs of hemorrhagic diathesis. Livers
affected acutely were enlarged and firm, with mottled discoloration and sometimes
a nutmeg appearance. In chronic intoxication, the liver was small, granular, and very
firm. A number of cases recovered after prolonged convalescence, and in these, there
was atrophy and fibrosis of the left and caudate lobes, and the right lobe was hyperplastic
and hemispheric. Histologically, in acute cases, there was widespread hemorrhagic
centrilobular necrosis and an unusual degree of intimal and subendothelial reaction
in sublobular and hepatic veins. The chronic lesion was dominated by extensive centrilobular
fibrosis, with obliterative changes in many central and sublobular veins. Sporadic
cases of portal hypertension in Greyhound dogs have been recognized where hepatic
veno-occlusive disease is the predominant lesion. In these cases, a history of feeding
meat treated with large amounts of nitrites or sulfites in an attempt to preserve
the color of fresh meat is common, but a causal relationship has not been established.
The hepatocellular changes are not specific for dimethylnitrosamine or other hepatotoxic
nitrosamines. Nitrosamines are metabolized in the liver and other tissues to reactive
alkyl groups that bind covalently to various macromolecules, especially guanine in
nucleic acids. Hepatotoxicity develops more slowly than in response to toxicants that
induce membrane peroxidation, as many hepatocytes with DNA damage undergo apoptosis
when stimulated to replicate. There is some megalocytosis, nuclear vesiculation and
nucleolar prominence, cytoplasmic intranuclear inclusions, and variable fatty change
and cytoplasmic bile accumulation. The typical effects of chronic experimental nitrosamine
hepatotoxicity, such as hepatocellular fibrosis, nodules, and neoplasms, have not
been evident in the accidental poisonings. Dimethylnitrosamine-induced hepatotoxicosis
in dogs has been proposed as a model of toxin-induced progressive hepatic disease
in this species.
Indospicine
Legumes of the genus
Indigofera
have long been known to contain the toxic amino acid indospicine (6-amidino-2-hexanoic
acid), which is a structural analogue of arginine, and which was shown in early experimental
work to be hepatotoxic for rats and other species. The dose rates necessary to cause
chronic liver injury in these species were, however, quite high, and field cases of
intoxications were only seen in cattle grazing I. spicata, which has the highest naturally
occurring concentrations of indospicine. In Australia, a serious outbreak of fatal
liver disease occurred in dogs that had been fed meat from horses that had been grazing
I. linnaei, a plant native to the arid zones of Australia that has been known to produce,
in horses, a chronic neurologic disorder known as “Birdsville horse disease.” The
disease in horses is not associated with liver damage, although these animals do accumulate
indospicine in most tissues, including muscle.
Dogs fed indospicine-contaminated meat for several weeks may develop progressive liver
damage. Affected livers may be small, firm, and pale, or they may be nodular because
of hypertrophy of surviving hepatocytes (eFig. 2-30A). Histologically, lesions begin
as vacuolation of a narrow band of centrilobular hepatocytes, followed shortly by
accumulation of mononuclear inflammatory cells in this zone and in the stoma of the
hepatic venules (eFig. 2-30B). Progression of the lesion is marked by scattered necrosis
of a widening zone of centrilobular hepatocytes, disorganization, vacuolation because
of fatty change, and accumulation of ceroid pigment in macrophages. Moderate centrilobular
fibrosis is seen in later stages, as well as pronounced canalicular cholestasis. By
this stage, affected animals begin to show icterus, inappetance, and depression. Bile
ductular proliferation is not a marked feature. Death is attended by the usual signs
of hepatoencephalopathy and tendency to bleed spontaneously. There is no evidence
of direct neurologic damage as is seen in horses. More recently, dogs ingesting canned
commercial camel meat developed serious and sometimes fatal liver disease. This may
be a concern for dogs with food allergies that are fed nonstandard protein sources
in special diets.
eFigure 2-30
A. Liver from a dog with indospicine toxicity with a zonal pattern of pallor.
(Courtesy R. Kelly.)
B. Mononuclear inflammatory infiltrates, biliary stasis, irregular vacuolation of
hepatocytes, and disorganization of hepatic plates in a dog.
This intoxication is remarkable by virtue of its unpredictability. Experimental intoxication
by pure indospicine has validly been shown to reproduce the naturally occurring intoxication
caused by horsemeat feeding; however, liver failure can be produced by either means
in only a small proportion of dogs so exposed. On the other hand, milder degrees of
liver damage are reliably produced by both methods. Thus it seems an idiosyncratic
response is superimposed upon a more consistent effect of the toxin; the nature of
the accompanying inflammatory response suggests that the former may be immunologically
mediated. This has yet to be established, as has the proposal that the mechanism of
the intoxication is the result of competitive inhibition of arginine.
Senna (Cassia) occidentalis
Ingestion of large amounts of seeds from Senna occidentalis in the family Leguminosae
can produce acute hepatic necrosis in cattle, pigs, goats, and horses. Hepatic injury
is characterized by lesions ranging from vacuolation, to scattered apoptosis and centrilobular
necrosis. In all species, except for horses, there is significant cardiac and skeletal
muscle injury and myoglobinuria that are the predominant lesions, rather than hepatic
injury. The toxic agent is not known currently.
Trifolium hybridum (alsike clover)
Alsike clover is a legume that grows well on wet clay soils in North America, where
it is sometimes responsible for outbreaks or single cases of cholangitis in horses
that consume it as pasture or hay. The toxic principle has not been isolated, but
it likely is in highest concentrations in the flowering stage used for hay. However,
there are evidently contributing factors because, although alsike clover is widespread
in cultivation, toxicity is rare. Toxicity usually occurs when alsike clover is a
major component of pastures or hay, but some horses are believed to graze it selectively
in a mixed pasture.
Clinical signs of poisoning initially include mild colic, ill-thrift, and anorexia,
with increasing occurrence of signs of hepatotoxicity with cholestasis. In some there
can be icterus and photodynamic dermatitis. Exposure to the plant can occur for a
year or more before signs of hepatic insufficiency develop, but cholestatic liver
disease can be detected biochemically before horses become ill. More severely affected
horses display neurologic disturbances, either excitement or mania in irregular episodes,
or long periods of extreme dullness, anorexia, apparent blindness, forced wandering,
head pushing, and yawning. Clinical problems related to hypoproteinemia, such as coagulopathy
or ascites, are not a feature of this disease, probably because most of the hepatocytes
are not directly affected.
The lesions of alsike clover poisoning are those of diffuse subacute to chronic biliary
hyperplasia (ductular reaction) and fibrosis, with the major lesions within and adjacent
to all intrahepatic biliary tracts (Fig. 2-102
). The liver may be enlarged, sometimes greatly so, or shrunken, pale, and tough or
rubbery in consistency. The surface is smooth, but its appearance is mottled. The
mottling is clear on the cut surface and is caused by bands of gray fibrous tissue,
distinctly visible to the naked eye, surrounding and compressing each lobule. Near
the margins of the liver and in some other areas, scar tissue may completely replace
the parenchyma. Areas of parenchymal atrophy may occur upstream of some occluded ducts.
Microscopically, there is pronounced proliferation of fibrous tissue in and around
the portal tracts associated with hyperplasia of well-formed bile ducts (ductular
reaction). Some areas have prominent neutrophil infiltration into ducts, but in many
areas this does not occur. In the early stages, the proliferation of bile ducts is
often greater than the proliferation of the fibrous tissue, but later the proportions
are reversed. The proliferating tissue extends slowly to connect adjacent portal triads,
circumscribing areas that correspond to conventional lobules. The fibrous tissue does
not permeate along the sinusoids, and there is gradual and uniform constriction of
the parenchyma.
Figure 2-102
Chronic cholangiohepatitis caused by alsike clover poisoning in a horse.
The distribution of liver lesions suggests that the harmful factor in alsike clover
is excreted into the bile in a form that damages the ducts. However, the biliary proliferation
and fibrosis is similar to that observed in areas of the liver that are drained by
chronically obstructed bile ducts, so it seems likely some of the later changes are
secondary to cholestasis. Agents that interfere with bile excretion have the potential
to interfere with elimination of various potentially injurious substances. Thus it
is possible that alsike clover ingestion might increase toxicity of other factors
in the forage.
Tephrosia cinerea
Tephrosia cinerea is a member of the Leguminosae family and is found in Brazil. Prolonged
ingestion causes chronic liver disease characterized clinically by wasting, and dramatic
ascites in affected sheep. Hydrothorax and hydropericardium can also be present. Livers
are pale with multiple small, ~1-mm diameter nodules. The livers are firm, and there
is gallbladder edema. Histologically, there is portal fibrosis and biliary hyperplasia
that is typically more severe in the subcapsular region. Most hepatocytes are enlarged
because of vacuolation. Scattered individual necrotic cells are present, and focally,
there is evidence of bile stasis. Inflammation is modest.
Brassica rapa
Turnip (Brassica rapa) or other Brassica family forage ingestion by cattle on the
North Island of New Zealand is incriminated as a cause of hepatogenous photosensitization
and biliary injury. The injury can mimic sporidesmin toxicity clinically with elevated
γ-glutamyl transferase (GGT) and phytoporphyrin plasma levels. Histologically, bile
ducts of smaller caliber than those injured by sporidesmin may be affected following
turnip forage ingestion.
Copper
The heavy-metal copper is an essential trace element that plays an important role
in numerous essential biological processes, including mitochondrial respiration (cytochrome
C oxidase), connective-tissue maturation cross-linking (lysyl oxidase), antioxidant
defense (superoxide dismutase), melanin synthesis (tyrosinase), iron metabolism (ceruloplasmin),
and neurotransmitter biosynthesis (dopamine β hydroxylase). However, copper is toxic
at excess concentrations because of its 2 redox states that can mediate free-radical
production, resulting in direct oxidation of cellular components. The liver is the
major organ involved in the regulation of copper levels, and homeostasis is maintained
by the balance of dietary intake and copper excretion via the bile. Dietary copper
is taken up by the enterocytes of the small intestine by dedicated transporters, divalent
metal transporter (DMT1) and copper transporter 1 (Ctr1), and conveyed in the portal
blood bound to carrier proteins ceruloplasmin and transcuprein, or in a nonspecific
fashion to albumin, to the liver for uptake. In the hepatocyte, copper is sequestered
in metallothionein or glutathione. Excretion into the bile or blood is regulated by
a series of specific chaperone proteins, such as the copper-transporting ATPase ATP7A.
Excess copper is stored in lysosomes. Copper-induced injury is produced by oxidative
processes that initially reduce available glutathione and eventually damages lipids,
proteins, and nucleic acids. Cellular metabolism, structural integrity, and energy
generation are affected. Hepatic copper toxicosis can result from a primary metabolic
defect in hepatic copper metabolism, altered hepatic biliary excretion of copper,
or from excess dietary intake of the element.
There is significant variation in species susceptibility to copper toxicosis. Sheep
as a species are most prone to copper poisoning, because of reduced biliary excretion
of copper. Some sheep breeds are more susceptible than others, reflecting differences
in the efficiency of intestinal absorption of copper, rather than differences in efficiency
of biliary excretion. Altered biliary excretion of copper in sheep does not appear
to be associated with alteration in the structure or expression of the gene for ATP7B,
the copper transporting P-type ATPase that is defective in Wilson disease, the human
autosomal recessive disorder of hepatic copper metabolism.
Excessive hepatic copper accumulation is important in chronic hepatitis in dogs, and
is discussed in further detail in the section Chronic hepatitis in dogs.
In cases of chronic hepatitis caused by copper accumulation, the liver is usually
small, often with an accentuated lobular pattern; severely affected livers are characterized
by architectural distortion, which ranges from a coarsely nodular texture to an end-stage
liver. Chronic hepatitis, depending on the duration of inflammation and injury, is
characterized by portal and periportal mononuclear cell inflammation and fibrosis
of portal areas that may extend into adjacent periportal areas of the lobule, leading
to the prominent lobular pattern. Small aggregates of pigmented macrophages, containing
copper and lipofuscin, surrounded by mononuclear inflammatory cells are a reliable
feature of copper excess. With progression, hyperplastic nodules and bridging fibrosis
develop.
In dogs and sheep, toxic amounts of copper can accumulate in the liver, although dietary
copper levels are not excessive by standards for other species. Copper poisoning does
occur in cattle and pigs, but in these species, it is because of abnormally high intake
of the element. Acute copper poisoning can occur following either the ingestion or
injection of excess copper, and animals deficient in vitamin E or molybdenum appear
especially susceptible to acute copper poisoning.
Chronic copper toxicosis in sheep occurs as a result of the presence of 3 environmental
factors acting alone or in concert. First, excessive copper intake may occur as a
result of contamination of water (naturally occurring or through the use of copper
piping or fixtures), pasture or prepared feed; the latter is difficult to avoid when
feed mills are preparing rations for different species and is probably partly responsible
for the observation that housed sheep are more prone to copper poisoning than animals
at pasture. Second, increased copper accumulation occurs as a result of increased
availability of dietary copper; this happens when dietary levels of molybdenum are
unusually low. Molybdenum, in the presence of sufficient sulfate, forms insoluble
complexes with copper in the gut and liver, making the copper biologically inert.
Subterranean clover growing on calcareous soils in southern Australia may be relatively
deficient in molybdenum, and in these areas, British breeds of sheep are known to
be more susceptible than Merinos to chronic copper poisoning. Other hepatotoxins constitute
the third environmental factor that predisposes sheep to outbreaks of chronic copper
poisoning. The most important of these are pyrrolizidine alkaloids (from Heliotropium
or Echium) in eastern Australia, and phomopsin from lupins in western Australia and,
possibly, South Africa.
The basis for chronic copper poisoning in sheep is the peculiar avidity of the liver
for copper, coupled with the very limited rate at which this species can excrete the
element in the bile. After intraportal injection of a copper isotope, practically
all the radioactivity is removed during the first passage through the liver. Most
of the copper is sequestered in hepatocellular lysosomes, where it does little damage
at concentrations of up to 200-300 µg/g dry weight. As the concentration rises, there
is presumably more interaction between other cell components and the copper. There
is some evidence that lysosomal membranes lose integrity and allow copper and lysosomal
hydrolases to damage the rest of the cytoplasm. By the time the liver copper concentration
has reached 300 µg/g or more, there is a histologically apparent increase in hepatocellular
turnover, with single hepatocytes undergoing apoptosis within a dense knot of neutrophils.
At still higher copper levels, the apoptotic rate increases, while all cells become
swollen and their nuclei vesicular. The mitotic rate increases, presumably to keep
pace with the accelerated loss of hepatocytes, and large macrophages appear in the
sinusoids and stromal spaces about the vessels. These cells contain eosinophilic or
somewhat brown, granular debris, which consists of copper-containing lipofuscins.
Sheep with liver copper concentrations >1,000 µg/g may be clinically and hematologically
normal, so long as the increasing mitotic rate produces enough new hepatocytes to
take up the copper released by dying cells. At this stage, however, there will be
elevated levels of liver-specific enzymes in the plasma. If the rate of hepatocellular
loss exceeds the capacity of the liver to phagocytose and sequester cell debris quickly,
the plasma copper levels can rise to levels that are high enough to damage circulating
erythrocytes, and intravascular hemolysis ensues. The effect of the hemolysis and
anemia on the liver is to accelerate the rate of hepatocellular necrosis; thus copper
enters the circulation at an increasing rate and acute copper toxic crisis is manifest.
The lethal clinical syndrome is then one of paroxysmal intravascular hemolysis and
liver failure, in which a sheep may pass from apparent good health to death within
~6 hours. Stresses, such as brief starvation, may also precipitate the crisis in susceptible
sheep, but the mechanisms involved are unknown. During the hemolytic crisis, some
of the copper is lost from the disintegrating liver; some passes into the urine, and
kidney copper concentration rises to 1,000 µg/g or more. Blood or kidney copper levels
therefore give a truer indication of a prior hemolytic crisis caused by chronic copper
poisoning than does elevation of liver copper alone.
The gross lesions of fatal chronic copper poisoning are those of acute copper toxicity.
The carcass is discolored by marked icterus, superimposed on which is the red color
imparted by free hemoglobin. Often, there is a brown hue as well, because a proportion
of the hemoglobin is oxidized to methemoglobin. The spleen is engorged, dark, and
soft. The kidneys are deep red-brown to black and the urine deep red, as a result
of hemoglobinuria with oxidation to methemoglobin, and concurrent icterus. The liver
is often slightly soft and swollen, and deep orange, but if the condition arises after
long-term liver injury, atrophy and fibrosis may be evident. The spectrum of liver
lesions can be complicated by other causes of liver necrosis, including hypoxia caused
by anemia, heart failure, and shock.
A breed of sheep from the Hebridean island of North Ronaldsay has apparently adapted
to a seaweed diet low in both copper and molybdenum but rich in zinc. Zinc is also
capable of interfering with copper uptake, and these sheep, although avoiding copper
deficiency, are exquisitely susceptible to chronic copper poisoning when transferred
to normal pasture. Hepatic disease associated with copper toxicity in this breed appears
to differ morphologically from other domesticated sheep breeds. Copper accumulation
begins in periportal hepatocytes, accompanied by a mixed inflammatory infiltrate and
cholangiolar proliferation. Characteristic pericellular fibrosis, initially confined
to the portal tracts and periportal zones, later extends to diffuse fibrosis and cirrhosis,
associated ultrastructurally with numerous hepatic stellate cells. North Ronaldsay
sheep have been proposed as a possible animal model for human non-Wilsonian hepatic
copper toxicosis and cirrhosis of infancy, and for investigation of copper-associated
hepatic fibrogenesis.
The events described in sheep also occur in chronic copper poisoning in pigs and cattle,
and acute intravascular hemolysis may be seen, especially in calves. Usually, however,
there is less of the acute terminal chain reaction in these species, and there is
more evidence of chronic liver damage with extensive portal fibrosis and biliary hyperplasia
within the triads.
Acute copper poisoning
is most often seen in ruminants after accidental administration of single large doses
of copper, by either the oral or parenteral routes. Iatrogenic copper toxicosis has
been induced by administering copper oxide boluses to neonatal calves, and by injection
with copper disodium edetate in weanling calves. Doses of 20-100 mg/kg can produce
acute poisoning in sheep. Copper toxicosis has also been reported in veal calves fed
milk replacer supplemented with various copper-containing hematinics. Affected animals
develop severe gastroenteritis, abdominal pain, diarrhea, and dehydration. The liver
lesion varies with chronicity of exposure, from nonspecific acute centrilobular necrosis,
to cholangiohepatitis with periportal fibrosis. Intravascular hemolysis may occur
if plasma copper levels are sufficiently elevated.
Acute bovine liver disease
Acute bovine liver disease is a poorly understood entity that occurs in southern Australia.
Outbreaks occur primarily in the spring and the autumn, and all ages of cattle can
be affected. Sudden death or photosensitization may occur. There is an association
with ingestion of Cynosurus echinatus (rough dog's tail). The characteristic histologic
lesion involves a periportal hepatocellular necrosis with bile duct proliferation
(eFig. 2-31). The presence of mycotoxin(s) produced by the fungus Dreschlera biseptata
growing in some plants has been suggested to be the principal toxicant or cofactor
in the syndrome.
eFigure 2-31
The acute bovine liver disease syndrome is characterized by periportal necrosis of
hepatocytes and bile duct proliferation.
(Courtesy R. Kelly.)
Further reading
Albretsen JC, et al. Cycad palm toxicosis in dogs: 60 cases (1987-1997). J Am Vet
Med Assoc 1998;213:99-101.
Allen JG, et al. The toxicity of Myoporum tetrandrum (boobialla) and myoporaceous
furanoid essential oils for ruminants. Aust Vet J 1978;54:287-292.
Allen JG, Hancock GR. Evidence that phomopsins A and B are not the only toxic metabolites
produced by Phomopsis leptostromiformis. J Appl Toxicol 1989;9:83-89.
Allen JG, Randall AG. The clinical biochemistry of experimentally produced lupinosis
in the sheep. Aust Vet J 1993;70:283-288.
Aslani MR, et al. In vitro detection of hepatocytotoxic metabolites from Drechslera
biseptata: a contributing factor to acute bovine liver disease? Aust J Exper Agric
2006;46:599-604.
Bandarra PM, et al. Trema micrantha toxicity in horses in Brazil. Equine Vet J 2010;42:456-459.
Bridges CH, et al. Kleingrass (Panicum coloratum L.) poisoning in sheep. Vet Pathol
1987;24:525-531.
Brum KB, et al. Intoxication by Vernonia rubricaulis in cattle in Mato Grosso do Sul.
Pesq Vet Bras 2002;22:119-128.
Casteel SW, et al. Chronic toxicity of fumonisin in weanling pigs. J Vet Diagn Invest
1993;5:413-417.
Cerqueira VD, et al. Colic caused by Panicum maximum toxicosis in Equidae in northern
Brazil. J Vet Diagn Invest 2009;21:882-888.
Cesar ASJ, et al. Toxic hepatopathy in sheep associated with the ingestion of the
legume Tephrosia cinerea. J Vet Diagn Invest 2007;19:690-694.
Collett MG. Bile duct lesions associated with turnip (Brassica rapa) photosensitization
compared with those due to sporidesmin toxicosis in dairy cows. Vet Pathol 2014;51:986-991.
Collett MG, et al. Photosensitisation, crystal-associated cholangiohepatopathy, and
acute renal tubular necrosis in calves following ingestion of Phytolacca octandra
(inkweed). N Z Vet J 2011;59:147-152.
Colvin BM, et al. Fumonisin toxicosis in swine: clinical and pathologic findings.
J Vet Diagn Invest 1993;5:232-241.
Cornick JL, et al. Kleingrass-associated hepatotoxicosis in horses. J Am Vet Med Assoc
1988;193:932-935.
Coulton MA, et al. Sporodesmin toxicosis in an alpaca. Aust Vet J 1997;75:136-138.
Cruz C, et al. Experimentally induced cholangiohepatopathy by dosing sheep with fractionated
extracts from Brachiaria decumbens. J Vet Diagn Invest 2001;13:170-172.
Edens LM, et al. Cholestatic hepatopathy, thrombocytopenia and lymphopenia associated
with iron toxicity in a Thoroughbred gelding. Equine Vet J 1993;25:81-84.
Ferguson D, et al. Survival and prognostic indicators for cycad intoxication in dogs.
J Vet Intern Med 2011;25:831-837.
FitzGerald LM, et al. Hepatotoxicosis in dogs consuming a diet of camel meat contaminated
with indospicine. Aust Vet J 2011;89:95-100.
Fu PP, et al. Pyrrolizidine alkaloids—genotoxicity, metabolism enzymes, metabolic
activation, and mechanisms. Drug Metab Rev 2004;36:1-55.
Garcia AF, et al. Comparative effects of lantadene A and its reduced metabolite on
mitochondrial bioenergetics. Toxicon 2010;55:1331-1337.
Glastonbury JRW, et al. A syndrome of hepatogenous photosensitization, resembling
geeldikkop, in sheep grazing Tribulus terrestris. Aust Vet J 1984;61:314-316.
Hamar DW, et al. Iatrogenic copper toxicosis induced by administering copper oxide
boluses to neonatal calves. J Vet Diagn Invest 1997;9:441-443.
Haywood S, et al. The greater susceptibility of North Ronaldsay sheep compared with
Cambridge sheep to copper-induced oxidative stress, mitochondrial damage and hepatic
stellate cell activation. J Comp Pathol 2005;133:114-127.
Holland PT, et al. Isolation of the steroidal sapogenin epismilagenin from the bile
of sheep affected by Panicum dichotomiflorum toxicosis. J Agric Food Chem 1991;39:1963-1965.
Hooser SB, et al. Actin filament alterations in rat hepatocytes induced in vivo and
in vitro by microcystin-LR, a hepatotoxin from the blue-green alga, Microcystis aeruginosa.
Vet Pathol 1991;28:259-266.
Howell JM, et al. Experimental copper and heliotrope intoxication in sheep: morphological
changes. J Comp Pathol 1991;105:49-74.
Ishmael J, et al. Experimental chronic copper toxicity in sheep. Biochemical and haematological
studies during the development of lesions in the liver. Res Vet Sci 1972;13:22-29.
Jago MV, et al. Lupinosis: response of sheep to different doses of phomopsin. Aust
J Exp Biol Med Sci 1982;60:239-251.
Jerrett IV, Chinnock RJ. Outbreaks of photosensitisation and deaths in cattle due
to Myoporum aff. Insulare R. Br. toxicity. Aust Vet J 1983;60:183-186.
Kellerman TS, et al. Photosensitivity in South Africa. VI. The experimental induction
of geeldikkop in sheep with crude steroidal saponins from Tribulus terrestris. Onderstepoort
J Vet Res 1991;58:47-53.
Koppang N. Toxic effect of dimethylnitrosamine in cows. J Natl Cancer Inst 1974;52:523-531.
Lockhart PJ, et al. Cloning, mapping and expression analysis of the sheep Wilson disease
gene homologue. Biochim Biophys Acta 2000;1491:229-239.
Mackie JT, et al. Lupinosis in yearling cattle. Aust Vet J 1992;69:172-173.
McLennan MW, Kelly WR. Cestrum parqui (green cestrum) poisoning in cattle. Aust Vet
J 1984;61:289-29.
Mendez MC, et al. Intoxication by Xanthium cavanillesii in cattle and sheep in southern
Brazil. Vet Hum Toxicol 1998;40:144-147.
Mercer HD, et al. Cassia occidentalis toxicosis in cattle. J Am Vet Med Assoc 1967;151:735-741.
Miles CO, et al. Identification of insoluble salts of the β-D-glucuronides of episarsasapogenin
and epismilagenin in the bile of lambs with alveld and examination of Narthecium ossifragum,
Tribulus terrestris, and Panicum miliaceum for sapogenins. J Agric Food Chem 1993;41:914-917.
Miles CO, et al. Photosensitivity in South Africa. VII. Chemical composition of biliary
crystals from a sheep with experimentally induced geeldikkop. Onderstepoort J Vet
Res 1994;61:215-222.
Munday R. Studies on the mechanism of toxicity of the mycotoxin, sporidesmin. V. Generation
of hydroxyl radical by sporidesmin. J Appl Toxicol 1987;7:17-22.
Nation PN. Hepatic disease in Alberta horses: a retrospective study of “alsike clover
poisoning” (1973-1988). Can Vet J 1991;32:602-607.
Oelrichs PB, et al. Isolation and identification of the toxic peptides from Lophyrotoma
zonalis (Pergidae) sawfly larvae. Toxicon 2001;39:1933-1936.
Oliveira-Filho JP, et al. Hepatoencephalopathy syndrome due to Cassia occidentalis
(Leguminosae, Caesalpinioideae) seed ingestion in horses. Equine Vet J 2013;45:240-244.
Patamalai B, et al. The isolation and identification of steroidal sapogenins in Kleingrass.
Vet Hum Toxicol 1990;32:314-318.
Peterson JE, et al. The toxicity of phomopsin for sheep. Aust Vet J 1987;64:293-298.
Puschner B, et al. Diagnosis of amanita toxicosis in a dog with acute hepatic necrosis.
J Vet Diagn Invest 2007;19:312-317.
Quinn JC, et al. Secondary plant products causing photosensitization in grazing herbivores:
Their structure, activity and regulation. Int J Mol Sci 2014;15:1441-1465.
Raposo JB, et al. Experimental intoxication by Myoporum laetum in sheep. Vet Hum Toxicol
1998;40:132-135.
Simola O, et al. Pathologic findings and toxin identification in cyanobacterial (Nodularia
spumigena) intoxication in a dog. Vet Pathol 2012;49:755-759.
Steffen DJ, et al. Copper toxicosis in suckling beef calves associated with improper
administration of copper oxide boluses. J Vet Diagn Invest 1997;9:443-446.
Stegelmeier BL. Pyrrolizidine alkaloid–containing toxic plants (Senecio, Crotalaria,
Cynoglossum, Amsinckia, Heliotropium, and Echium spp.). Vet Clin North Am Food Anim
Pract 2011;27:419-428.
Stegelmeier BL, et al. Pyrrole detection and the pathologic progression of Cynoglossum
officinale (houndstongue) poisoning in horses. J Vet Diagn Invest 1996;8:81-90.
Sullivan JM, et al. Copper toxicosis in veal calves. J Vet Diagn Invest 1991;3:161-164.
Tan ET, et al. Determination of hepatotoxic indospicine in Australian camel meat by
ultra-performance liquid chromatography-tandem mass spectrometry. J Agric Food Chem
2014;62:1974-1979.
Tapia MO, et al. An outbreak of hepatogenous photosensitization in sheep grazing Tribulus
terrestris in Argentina. Vet Hum Toxicol 1994;36:311-313.
Tegzes JH, Puschner B. Amanita mushroom poisoning: efficacy of aggressive treatment
of two dogs. Vet Hum Toxicol 2002;44:96-99.
Thiel C, et al. The enterohepatic circulation of amanitin: kinetics and therapeutical
implications. Toxicol Lett 2011;203:142-146.
Tokarz D, et al. Amanitin toxicosis in two cats with acute hepatic and renal failure.
Vet Pathol 2012;49:1032-1035.
Van Der Lugt JJ, et al. Experimentally-induced Cestrum laevigatum (Schlechtd.) poisoning
in sheep. Onderstepoort J Vet Res 1992;59:135-144.
Voss KA, Riley RT. Fumonisin toxicity and mechanism of action: overview and current
perspectives. Food Safety 2013;1:2013006-2013006.
Wisløff H, et al. Accumulation of sapogenin conjugates and histological changes in
the liver and kidneys of lambs suffering from alveld, a hepatogenous photosensitization
disease of sheep grazing Narthecium ossifragum. Vet Res Commun 2002;26:381-396.
Yee MM, et al. Amanitin intoxication in two beef calves in California. J Vet Diagn
Invest 2012;24:241-244.
Hyperplastic and Neoplastic Lesions of the Liver and Bile Ducts
Remarkably, the occurrence of fatal liver malignancies is rather uncommon in aged
dogs and cats; this suggests that they are either less exposed, or they are more resistant
to the etiologic agents responsible for liver neoplasms in humans. Domestic species
are not subject to significant chronic oncogenic viral infections, such as the hepatitis
B and hepatitis C viruses that account for the majority of human hepatocellular carcinoma.
Ectopic, metaplastic, and hyperplastic lesions
Ectopic and metaplastic lesions in the gallbladder have been reported. Ectopic tissue
includes hepatocyte nodules attached to or within the wall, and pancreatic islands,
which may include islets of Langerhans. Gastric ectopia, readily distinguishable by
the presence of chief and parietal cells, may form plaques or large polyps. Ectopia
of cardiac or pyloric mucosa is mimicked by metaplastic changes.
Nodular hyperplasia of hepatocytes is common in old dogs, but rare in other species.
The lesions in dogs do not have a breed or sex predisposition, but their incidence
increases sharply with age. Hyperplastic nodules typically develop in livers of normal
mass, whereas regenerative nodules arise as a result of compensatory hyperplasia of
surviving hepatocytes in a background of hepatic injury, atrophy, and fibrosis (see
Responses of the liver to injury).
Nodular hyperplasia often occurs as multiple randomly distributed masses throughout
the lobes. The grossly visible nodules are spherical, well circumscribed, varying
in size from 2 mm to 3 cm or more, and may bulge from the capsular surface or be entirely
hidden within the parenchyma (Fig. 2-103
). They may be sharply distinct from the surrounding parenchyma because of color differences,
either lighter than the surrounding liver because of increased lipid or glycogen content
within hepatocytes comprising the nodule, or darker because the sinusoidal vessels
are distended with blood. Some nodules are the same color as the surrounding tissue
and can only be identified by examination of a washed or blotted slice under a strong
light.
Figure 2-103
Nodular hyperplasia in the liver of a dog.
The larger hyperplastic nodules grow expansively; they do not induce a fibrous capsule,
but they can compress the surrounding parenchyma. The hepatocytes comprising nodular
hyperplasia are often phenotypically different from those in adjacent parenchyma (Fig.
2-104
). Old canine livers often have microscopic focal hyperplasia of similarly altered
hepatocytes that can be distinguished from the surrounding parenchyma, but they evidently
grow more slowly and do not compress their boundaries. These can be regarded as examples
of the numerous atypical focal hyperplasias that arise as altered foci, nodules, plaques,
or polyps in various tissues of old dogs. In more prominent paler nodules, hepatocytes
can be enlarged and vacuolated with lipid or glycogen accumulation. Mitotic figures
are uncommon. The sinusoids of the nodules are often dilated, and foci of hematopoiesis
are occasionally observed. Necrosis and hemorrhage in hyperplastic nodules are rare.
Figure 2-104
Nodular hyperplasia in the liver of a dog. These nodules have a discrete border with
adjacent parenchyma and often are vacuolated.
The significance of hyperplastic nodules in old dogs is usually negligible. After
acute liver necrosis, these atypical hepatocytes within pre-existing hyperplastic
foci and nodules may have phenotypic alterations that confer a survival advantage,
and can survive and flourish as larger nodules amid regions of necrosis, so in these
instances, they can be beneficial. Although hyperplastic nodules rarely progress to
lesions with detrimental impact, they are often found during liver imaging, laparotomy,
or biopsy and need to be differentiated from other focal lesions of more significance.
Because they are atypically differentiated, they are the potential source of secreted
products of clinical or diagnostic significance, but their potential in this regard
is unexplored. The hallmark of nodular hyperplasia, in contrast to the hepatic adenoma
and hepatocellular carcinoma, is that it largely retains normal liver architecture,
including a modified lobular structure with recognizable central veins and portal
triads. However, this feature may be difficult to appreciate in small biopsies. The
biological distinction between atypical hyperplasia and neoplasia is sometimes equivocal;
in the latter, continued growth that is independent of promoting influences is responsible
for enlargement, and the designation of adenoma should apply to lesions that are enlarging.
Cystic mucinous hyperplasia
of the mucus-producing glands in the mucosa of the gallbladder has been reported as
an incidental lesion in dogs and sheep. These cystic hyperplastic nodules are often
numerous, may be sessile or polypoid, and contain mucin (Fig. 2-105
).
Figure 2-105
Cystic mucinous hyperplasia of the mucosa of the gallbladder in a dog.
Dilation of the gallbladder with accumulated mucoid secretion (gallbladder mucocele)
has been reported in dogs and infrequently in cats. Histologically, affected gallbladders
display proliferated mucosa with characteristic projecting fronds of epithelium that
extend into the lumen of the gallbladder, as well as formation of mucus-filled cysts.
Severely affected gallbladders are distended with abnormal semisolid accumulations
of mucus or other secretions, and the wall may undergo ischemic necrosis and rupture
(Fig. 2-106
). Bile-laden mucus may also extend into the cystic, hepatic, and common bile ducts,
resulting in variable degrees of extrahepatic biliary obstruction. The etiology of
this condition is unknown, and may be related to decreased gallbladder motility, bile
stasis, and altered bile composition and viscosity.
Figure 2-106
Gallbladder mucocele in a dog.
Mucosal hyperplasia in the large bile ducts is frequently observed in the long-standing
mild cholangiohepatitis of fluke infestation. The hyperplasia is of microscopic dimensions,
but in some instances, it appears histologically to be atypical. Localized, polypoid
foci of cystic hyperplasia are specific changes in cattle poisoned by highly chlorinated
naphthalene.
Hepatocellular tumors
Primary epithelial neoplasms of the liver include tumors of either hepatocellular
or cholangiocellular origin, and they are uncommon in most species, representing <1%
of all neoplasms in cats and dogs. Pot-bellied pigs are a possible exception. Proportionately,
hepatocellular neoplasms are more common in dogs, whereas cholangiocellular tumors
predominate in cats. Unlike the situation in humans, there are no clear associations
in domestic animals between hepatocellular tumors and viruses, chemical carcinogens,
mycotoxins, or drugs, such as synthetic steroids. With the exception of the occasional
association of liver tumors with certain flukes or tapeworm larvae, and the rare adenoma
seen in the bovine liver with chronic pyrrolizidine poisoning, hepatobiliary tumors
in domestic animals are not obvious successors to recognized antecedent liver diseases.
Hepatocellular adenomas are benign neoplasms of hepatocytes. Hepatocellular adenomas
are reported in dogs, cats, cattle, sheep, and pigs, but likely occur in all species.
They are rarely of any clinical significance. Hepatocellular adenomas are typically
solitary, but they can be multiple. They range from 2-12 cm in diameter and are roughly
spherical masses. Typically, they are well demarcated from the adjacent parenchyma
because of the compression caused by their expanding growth, but not encapsulated.
Hepatocellular adenomas often bulge from the capsular surface, or they can be pedunculated,
and in some instances, they may be found entirely within the hepatic parenchyma. The
color of hepatocellular adenomas varies from yellow-brown to dark mahogany red. Paler
adenomas contain lipid or glycogen, and these lesions often have a soft and friable
consistency compared to normal liver (Fig. 2-107
).
Figure 2-107
Hepatocellular adenoma in a dog.
Histologically, the hepatocytes in hepatic adenomas do not differ markedly from normal
hepatocytes or those in hyperplastic nodules, and often contain lipid, glycogen, or
occasionally protein secretory vacuoles. Minimal anisocytosis and increased basophilia
of the cytoplasm and prominence of nucleoli can be seen, but mitotic figures are uncommon.
Cells are arranged in cords or trabeculae that may be several cells thick, but the
width tends to be consistent. There is a discrete well-defined border with adjacent
parenchyma (Fig. 2-108
). Adenomas lack normal lobular architecture. Essentially all of the blood supplying
adenomas comes from the hepatic artery. As a consequence, rather than portal tracts,
isolated arteries, bile ducts, and hepatic veins can be found coursing through the
mass. Usually, no more than a single portal tract may be present, likely entrapped
by the expanding hepatocyte population. Extramedullary hematopoietic foci may sometimes
be found in the adenomas, as well as ectatic vascular spaces. Focal necrosis may occur.
Figure 2-108
Histologic appearance of a canine hepatocellular adenoma. Compression of the adjacent
parenchyma is apparent, and a capsule is evident at this site.
Hepatocellular carcinomas are uncommon, but occur in all veterinary species; they
may be single and massive, nodular or diffuse. Several features suggest malignancy,
including the absence of pedunculation or clear demarcation from the adjacent parenchyma
and the presence of varied coloration of the cut surface produced by hemorrhage and
necrosis (eFig. 2-32). Venous invasion is typical of the hepatocellular tumors, and
intravascular spread may extend to the large hepatic veins and vena cava. Carcinomas
occasionally penetrate the capsule to implant on the peritoneum. Metastasis is uncommon
in all domestic species but, when present, may be evident in the hepatic lymph node
most often. Hematogenous metastases occur first in the lungs. Spontaneous rupture
is common and may cause fatal blood loss. This consequence is a much more frequent
cause of morbidity and mortality than metastatic disease. Some hepatocellular carcinomas
in cattle are scirrhous, hard, and white. Paraneoplastic hypoglycemia may occur in
animals with hepatocellular carcinoma.
eFigure 2-32
Hepatocellular carcinoma in a dog.
Hepatocellular carcinomas may be quite variable in histologic appearance, and multiple
patterns may occur within a single neoplasm. Trabecular carcinomas are the most common
histologic pattern (Fig. 2-109
). Neoplastic cells resemble normal liver, and grow in irregularly thick plates or
trabeculae that vary from few to many cells thick. Necrosis may be present, and sinusoids
may be dilated, and there may be large ectatic blood-filled spaces. Other patterns
include pseudoglandular (adenoid) carcinomas, characterized by lobular arrangement
of neoplastic hepatocytes within scant connective-tissue stroma, and solid carcinomas,
characterized by solid sheets of poorly differentiated, often pleomorphic cells that
do not form sinusoids (Fig. 2-110A, B
). A scirrhous variant has been described in dogs, characterized by multiple foci
of cytokeratin 19–positive ductular structures embedded in abundant fibrous stroma
(Fig. 2-111A, B
). A feature of some hepatocellular tumors is the presence of giant cells, quite conspicuous
by virtue of a large nucleus, multilobed nuclei, or multiple nuclei. Mitotic figures
are more frequent in carcinomas than adenomas, but may not be a prominent feature
of well-differentiated carcinomas. Immunohistochemical staining using antibodies that
bind a hepatocyte specific antigen, HepPar1, identifies a large proportion of canine
and feline hepatocellular carcinomas. However, more aggressive forms of canine hepatocellular
carcinoma may not stain with HepPar1, but instead >5% of the cells can be stained
with antibodies that recognize cytokeratin 19, a marker of hepatic progenitor cells,
immature hepatocytes, and biliary epithelial cells. These canine hepatocellular carcinomas
have an aggressive growth pattern, including a markedly increased incidence of intrahepatic
and extrahepatic metastasis.
Figure 2-109
Trabecular hepatocellular carcinoma from a dog.
Figure 2-110
A. A solid pattern in a poorly differentiated hepatocellular carcinoma from a dog.
B. Invasion at the margin of a hepatocellular carcinoma in the liver of a dog.
Figure 2-111
A. The scirrhous variant of hepatocellular carcinoma has areas of ductular proliferation
and abundant connective tissue within areas of neoplastic hepatocytes. B. Ductular
structures staining with cytokeratin 19.
Hepatoblastomas, rare benign tumors putatively originating from primitive hepatic
precursor cells, have been reported most often in horses, but also in sheep, dogs,
cats, and a llama. Hepatoblastomas in humans are neoplasms of infancy and childhood.
Cases in domestic species have been reported in both young and adult animals, but
are most frequently reported in foals. Hepatoblastomas are typically single, firm,
lobulated masses with areas of necrosis and hemorrhage. Compression of adjacent parenchyma
is evident and, on occasion, invasion can be seen. The histologic patterns of hepatoblastomas
can be epithelial, either fetal or embryonal, or mixed epithelial and mesenchymal.
Fetal hepatoblastomas are composed of large, polygonal cells, approximately the size
of adult hepatocytes, with round to oval nuclei and granular to vacuolated, eosinophilic
to amphophilic cytoplasm (Fig. 2-112A
). Embryonal hepatoblastomas form ribbons or rosettes of smaller basophilic cells
with scant cytoplasm (Fig. 2-112B). Mixed epithelial-mesenchymal hepatoblastomas contain
variable amounts of fibrous connective tissue or other mesenchymal tissues, including
cartilage or bone, in addition to epithelial cells. Combinations of these patterns
are common. Portal tracts are absent. Immunohistochemical staining for α-fetoprotein
is typically positive, and HepPar-1 staining is usually absent, supporting the view
that they arise from precursor cells.
Figure 2-112
A. Hepatoblastoma with a fetal pattern from a foal. B. Hepatoblastoma with an embryonal
pattern from a foal.
Cholangiocellular tumors
Cholangiocellular adenomas are uncommon benign neoplasms of biliary epithelium. Cholangiocellular
adenomas are usually solitary, pale gray to white, and well-circumscribed roughly
spherical masses that tend to grow by expansion. They may distend the normal outline
of the liver, although they can occur as intrahepatic masses. Cholangiocellular adenomas
are composed of tubules lined with a single layer of well-differentiated biliary epithelium
and a moderate amount of intervening stroma. The tubules may have narrow lumens, and
there can be variable amounts of stroma within the mass (Fig. 2-113
). Adjacent hepatocytes are usually compressed at the margins, but are not found between
tubules. The tubules contain a clear watery to viscous fluid. Cuboidal or flattened
neoplastic biliary epithelial cells have a moderate amount of pale eosinophilic cytoplasm.
Nuclei are round to oval, vesicular, and oriented centrally. Nucleoli are small or
inapparent. There are typically no mitotic figures.
Figure 2-113
Biliary adenomas are composed of uniform arrays of well-differentiated biliary epithelium
forming uniform small tubules.
Extrahepatic cholangiocellular adenomas are rare, with the exception of gallbladder
adenomas reported in abattoir surveys of cattle and rarely in domestic carnivores
(Fig. 2-114
). The smaller specimens may be solid on cut surface and white, but the large specimens,
and many of the small ones, are cystic.
Figure 2-114
Gallbladder adenoma in a dog.
(Courtesy J. Tobias.)
Lesions formerly termed biliary cystadenomas, a subtype commonly seen in cats and
occasionally in dogs, are most likely developmental anomalies of the embryonal biliary
tree, termed ductal plate anomalies, which is more clearly appreciated when biliary
cysts are coincident with cystic renal developmental lesions, although either lesion
may occur independently. The ductal plate anomalies can be multilocular and lined
by bile duct epithelia that may be flattened by pressure, or in some areas papillary.
The septal stroma is collagenous. Their development later in life may be the result
of progressive secretion by the lining epithelial cells, creating cysts only when
sufficient fluid has been produced. Entrapped hepatocytes are often present.
Cholangiocarcinomas (cholangiocellular carcinomas) are reported in dogs, cats, sheep,
cattle, horses, and goats. Affected livers are usually otherwise normal, with no suggestion
as to an underlying cause, although there are associations with chronic fluke infestations
in humans and rarely in carnivores.
Cholangiocellular tumors can usually be distinguished from the hepatocellular variety
by their multiplicity, firmness, pale beige color produced by more or less abundant
stroma, and the typical umbilicate appearance of those that involve the capsule (Fig.
2-115
). The central depressed area can be the result of necrosis or cavitation-associated
collapse of tumor vessels in the central parts of the tumor nodules. Even multiple
nodules may not cause much enlargement of the liver. In dogs and cats, the tumors
are almost always multiple or diffuse. The multiple nodules of tumor might represent
intrahepatic lymphogenous metastases, but the possibility of multicentric origin must
be entertained. Hematogenous metastases are unusual, but metastases to the regional
nodes are common. In cats especially, there is a tendency to invade Glisson's capsule
and implant on the peritoneum; the diffuse variety may cause great enlargement, although
with retention of shape.
Figure 2-115
A cholangiocarcinoma in the liver of a dog. Umbilication is prominent.
(Courtesy J.L. Caswell.)
Microscopically, cholangiocellular carcinomas form ductules and acini, and sometimes
papillary formations within the lumen of the neoplastic ducts (Fig. 2-116
). The cells are cuboidal or columnar, with a small amount of clear or slightly granular
cytoplasm. The nuclei are small and fairly uniform, and nucleoli are not prominent.
Mitotic figures are often abundant. The tubules do not contain bile, but in well-differentiated
specimens may contain mucins. The epithelial components are separated by fibrous connective-tissue
stroma, which may have pronounced collagen deposition, the so-called scirrhous response
that gives the tumor a firm texture. In poorly differentiated cholangiocarcinomas,
pleomorphic to anaplastic cells can be observed. It is not uncommon to encounter areas
of poorly differentiated cells within a mass that is predominantly well differentiated.
Cholangiocellular carcinomas have a highly invasive growth pattern, and frequently
metastasize to hepatic lymph nodes, lungs, and the peritoneal cavity.
Figure 2-116
Cholangiocarcinoma from a cat with characteristic crude acini and tubules separated
by abundant connective tissue.
Primary intrahepatic cholangiocellular carcinoma can be difficult, and often impossible,
to distinguish from metastatic adenocarcinomas, especially those of pancreatic or
mammary epithelial origin. Mucus secretion and intrasinusoidal permeation are more
typical of biliary origin. Distinction from hepatocellular carcinoma of adenoid pattern
may be assisted by demonstration of mucin, abundant mitotic figures, and presence
of a prominent connective-tissue stroma.
Extrahepatic cholangiocellular carcinoma (or biliary cholangiocarcinoma) of the gallbladder
or extrahepatic bile ducts is much less frequent than the intrahepatic form, but has
been reported in dogs, cats, cattle, and swine.
Mixed hepatocellular and cholangiocellular carcinomas
Rare hepatic carcinomas have the histologic and cytologic characteristics of both
hepatocellular carcinoma and cholangiocellular carcinoma. These tumors likely arise
from bipotential hepatic progenitor cells. The hepatocytic nature of some tumor cells
can be confirmed with the monoclonal antibody HepPar-1, and cells of biliary phenotype
can be stained by immunohistochemistry using antibodies that bind to cytokeratin 7,
a typical intermediate filament of biliary epithelium (Fig. 2-117A-C
).
Figure 2-117
A. Mixed hepatocellular and biliary carcinomas contain neoplastic cell populations
with both biliary and hepatocellular phenotypes. H&E. B. HepPar1-stained cells. C.
Cytokeratin 7–stained cells.
Hepatic carcinoids
Hepatic carcinoids, presumably arising from the diffuse neuroendocrine cell population
found among the biliary epithelium and possibly within the hepatic parenchyma, have
been reported in dogs, cats, and cattle. They may arise within the liver, in the extrahepatic
bile ducts or within the gallbladder. The gross pathologic appearance of carcinoids
is typically that of disseminated pale gray to tan small masses within the liver,
but on some occasions, only a single mass is formed (eFig. 2-33). Like other neuroendocrine
tumors, hepatic carcinoids typically form nests of cells separated by a fine fibrovascular
stroma. The cells are oval to spindle shaped, and may form a rosette or pseudolobular
pattern (Fig. 2-118
). Mitotic figures are usually frequent. Argyrophilic cytoplasmic granules may be
detected by silver impregnation stains; more precise identification of carcinoids
may require immunohistochemical stains for neurosecretory products, such as neuron-specific
enolase or serotonin. These are aggressive neoplasms, with frequent intrahepatic spread,
and metastasis to local lymph nodes, peritoneum, and lung.
Figure 2-118
Hepatic carcinoid from a cat with characteristic islands and rosettes separated by
fine fibrovascular stroma and characteristic invasive behavior.
eFigure 2-33
Hepatic carcinoid from a cat.
Mesodermal tumors
Primary mesenchymal tumors of the liver are quite uncommon. Of the mesenchymal tumors,
primary hemangiosarcomas are likely the most frequent. Primary hepatic hemangiosarcomas
occur in dogs, cats, cattle, and sheep. Hemangiosarcoma of the liver should always
be considered metastatic until proven otherwise by diligent search. Some of these
tumors are solitary, large, and gray-white, with scattered hemorrhagic areas, and
others are ill defined and cavernous (Fig. 2-119
). The latter may rupture into the peritoneal cavity to produce severe hemorrhage.
Microscopically, hepatic hemangiosarcomas resemble those found in other sites; however,
it may be impossible to find malignant cells in or lining cavernous areas. At the
margins of the tumor, there is a distinctive pattern or growth in which small, solid
nodules of malignant cells may be found, or these cells can be found forming capillary
structures or invading along pre-existing sinusoids. The latter phenomenon is particularly
characteristic. As the cells invade along the sinusoids, perhaps in single file, they
initially produce little distortion of the hepatic cords. Behind them, the sinusoids
are spread widely apart, and individual hepatocytes or portions of cords are isolated
and appear to be floating freely, surrounded by a thin layer of connective tissue
and neoplastic cells. Differential diagnoses other than metastatic lesions include
the syndrome of telangiectasia in Pembroke Welsh Corgi dogs, telangiectatic lesions
in older animals, and vascular hamartomas in cattle. Hepatic hemangiomas have been
reported in dogs and a pig.
Figure 2-119
Hepatic hemangiosarcoma in the liver of a dog. Careful examination is required to
distinguish primary from metastatic disease.
Leiomyomas are occasionally observed in the gallbladder of dogs and cattle. Leiomyosarcomas
and fibrosarcomas have been reported in the liver of cats and dogs, hemangiopericytoma
and fibrosarcoma in cattle, and a single case of botryoid-type embryonal rhabdomyosarcoma
in a cat. Other rare mesenchymal tumors reported in either dogs or cats include lymphangioma,
plasmacytoma, osteosarcoma, nerve sheath tumor, liposarcoma, and chondrosarcoma.
The myelolipoma is an unusual tumor that develops in the livers of domestic cats and
wild felids. The tumor develops as multiple growths, 0.5-5 cm diameter, in one or
more lobes; if they project above the surface of the liver, they are irregularly nodular.
Myelolipomas are friable, and yellow to orange because of their high fat content.
The neoplasm is composed of normal-appearing, mature adipocytes with a variable admixture
of myeloid cells, including both mature and immature cells of the granulocytic, erythrocytic,
and megakaryocytic series (Fig. 2-120
). In captive wild cats, similar lesions have been seen in the spleen. These were
judged to be separate developments of the same process. Metastasis to other organs
has not been reported.
Figure 2-120
Myelolipoma of the liver of a dog.
Metastatic neoplasms
The liver is particularly “fertile soil,” as metastatic lesions markedly outnumber
primary hepatic neoplasms. Metastatic solid neoplasms may be multiple, but are usually
not numerous and usually do not elicit clinical or biochemical evidence of hepatic
injury or dysfunction. Malignancies arriving via the portal vein, such as pancreatic
or gastric carcinoma or hemangiosarcomas, may practically replace the liver before
producing clinical signs, one of which may be icterus caused either by extrahepatic
bile duct obstruction, or by intrahepatic cholestasis, or both. In dogs, the most
common metastatic hematopoietic, mesenchymal, and epithelial neoplasms are lymphoma,
hemangiosarcoma, and pancreatic carcinoma, respectively. Some of the carcinomas (e.g.,
thyroid, mammary), melanoma, and sarcomas from more remote sites also metastasize
to the liver via the lungs and hepatic artery, and some (e.g., ovarian carcinoma,
mesothelioma) implant on the capsular surface from within the peritoneal cavity.
The hepatic perisinusoidal and periportal compartments are hospitable to various hematopoietic
neoplastic cell types. Accordingly, malignant histiocytosis, myeloid and erythroid
leukemias, and mast cell tumors often localize in the liver along hepatic sinusoids.
Some lymphomas, histiocytic sarcomas (Fig. 2-121
), plasmacytomas (Fig. 2-122
), and mast cell tumors also produce solid sarcoma masses in the liver as primary
or metastatic lesions. Lymphoma in the liver is common, especially when the spleen
is involved, and occasionally, the liver appears to be the major or primary site affected.
Typically, lymphoma is most apparent in the portal tracts, and the connective tissue
surrounding the central veins can also be infiltrated, but late in the course of disease,
the sinusoids are affected (eFig. 2-34). There are occasional exceptions to this pattern.
Hepatosplenic γδ T-cell lymphomas (also CD3+ and CD11d+) in dogs demonstrate preferential
involvement of the liver and spleen, and neoplastic lymphocytes are most common within
the sinusoids. Hepatocytotrophic lymphoma is a T-cell lymphoma variant (CD3+, CD11d−)
that invades hepatocytes. Diffuse infiltration of the liver in myeloproliferative
disorders and mast cell leukemia may also cause extreme enlargement of the organ;
the infiltrates localize preferentially in and around the sinusoids (Fig. 2-123
). Hepatic infiltration may be in discrete nodules 2 cm or more in size, particularly
lymphoma in horses, but it is usually diffuse in the connective tissues of the portal
triads.
Figure 2-121
Histiocytic sarcoma in the liver of a dog.
Figure 2-122
Primary hepatic plasmacytoma in the liver of a dog.
Figure 2-123
Monomyelocytic leukemia cells typically array along the hepatic sinusoids as seen
in this dog.
eFigure 2-34
Lymphoma typically infiltrates the stroma of portal tracts and hepatic venules as
seen in this dog.
Melanomas and hemangiosarcomas have characteristic gross features in the liver, but
most metastatic tumors cannot be distinguished by their gross appearance. Sarcomas
do tend to form a few large, smooth-surfaced nodules, and carcinomas do tend to form
more nodules and to be umbilicate when in contact with Glisson's capsule. Hemangiosarcomas
come from the spleen, usually, and may virtually replace the liver with small, blood-filled
caverns. Their microscopic appearance is the same as that of the primary tumors.
A non-neoplastic condition, hepatic splenosis, a rare condition in which normal splenic
tissue becomes implanted into the liver, can be confused with metastatic disease.
Normal spleen enters the liver, often via the portal vein after surgery or trauma
to the spleen and multiple soft red masses up to ~4 cm in diameter can develop. Histologically,
the masses resemble splenic tissue.
Further reading
Beeler-Marfisi J, et al. Equine primary liver tumors: a case series and review of
the literature. J Vet Diagn Invest 2010;22:174-183.
Bergman JR. Nodular hyperplasia in the liver of the dog: an association with changes
in the Ito cell population. Vet Pathol 1985;22:427-438.
Bettini G, Marcato PS. Primary hepatic tumours in cattle. A classification of 66 cases.
J Comp Pathol 1992;107:19-34.
Bosje JT, et al. Polycystic kidney and liver disease in cats. Vet Q 1998;20:136-139.
Cantile C, et al. Hepatoblastoma in a foal. Equine Vet J 2001;33:214-216.
Charles JA, et al. Morphological classification of neoplastic disorders of the canine
and feline liver. In: Rothuizen J, et al., editors. WSAVA Standards for Clinical and
Histological Diagnosis of Canine and Feline Liver Diseases. Philadelphia: Saunders
Elsevier; 2006. p. 117-124.
Desmet VJ. Congenital diseases of intrahepatic bile ducts: variations on the theme
“ductal plate malformation.” Hepatol 1992;16:1069-1083.
Fabry A, et al. Nodular hyperplasia of the liver in the beagle dog. Vet Pathol 1982;19:109-119.
Fry MM, et al. Hepatosplenic lymphoma in a dog. Vet Pathol 2003;40:556-562.
Gamlem H, et al. Canine vascular neoplasia—a population-based clinicopathologic study
of 439 tumours and tumour-like lesions in 420 dogs. APMIS Suppl 2008;125:41-54.
Haddad JL, Habecker PL. Hepatocellular carcinomas in Vietnamese pot-bellied pigs (Sus
scrofa). J Vet Diagn Invest 2012;24:1047-1051.
Haines VL, et al. Adenocarcinoma of the hepatopancreatic ampulla in a domestic cat.
Vet Pathol 1996;33:439-441.
Knostman KA, et al. Intrahepatic splenosis in a dog. Vet Pathol 2003;40:708-710.
Lawrence HJ, et al. Nonlymphomatous hepatobiliary masses in cats: 41 cases (1972 to
1991). Vet Surg 1994;23:365-368.
Minkus G, Hillemanns M. Botryoid-type embryonal rhabdomyosarcoma of liver in a young
cat. Vet Pathol 1997;34:618-621.
Moore FM, Thornton GW. Telangiectasia of Pembroke Welsh corgi dogs. Vet Pathol 1983;20:203-208.
Mueller POE, et al. Antemortem diagnosis of cholangiocellular carcinoma in a horse.
J Am Vet Med Assoc 1992;201:899-901.
Neu SM. Hepatoblastoma in an equine fetus. J Vet Diagn Invest 1993;5:634-637.
Patnaik AK, et al. Canine bile duct carcinoma. Vet Pathol 1981;18:439-444.
Patnaik AK, et al. Canine hepatic carcinoids. Vet Pathol 1981;18:445-453.
Patnaik AK, et al. Canine hepatocellular carcinoma. Vet Pathol 1981;18:427-438.
Patnaik AK, et al. Hepatobiliary neuroendocrine carcinoma in cats: a clinicopathologic,
immunohistochemical, and ultrastructural study of 17 cases. Vet Pathol 2005;42:331-337.
Pollock S, Wagner BM. Hemangioma of the liver in a dog. Vet Med Small Anim Clin 1972;67:863-867.
Post G, Patnaik AK. Nonhematopoietic hepatic neoplasms in cats: 21 cases (1983-1988).
J Am Vet Med Assoc 1992;201:1080-1082.
Rodríguez F, et al. Cholangiocarcinoma in a goat. Vet Rec 1996;139:143-144.
Shiga A, et al. Hepatoblastoma in a dog. J Vet Med Sci 1997;59:1167-1170.
Tanimoto T, Ohtsuki Y. Hepatic haemangioma in a pig. Vet Rec 1992;131:176-177.
van den Ingh TS, Rothuizen J. Congenital cystic disease of the liver in seven dogs.
J Comp Pathol 1985;95:405-414.
van den Ingh TSGAM, et al. Morphological classification of biliary disorders of the
canine and feline liver. In: Rothuizen J, et al., editors. WSAVA Standards for Clinical
and Histological Diagnosis of Canine and Feline Liver Diseases. Philadelphia: Saunders
Elsevier; 2006. p. 61-76.
van Sprundel RG, et al. Classification of primary hepatic tumours in the dog. Vet
J 2013;197:596-606.
Zini E, et al. Paraneoplastic hypoglycemia due to an insulin-like growth factor type-II
secreting hepatocellular carcinoma in a dog. J Vet Intern Med 2007;21:193-195.