- Record: found
- Abstract: found
- Article: found
β-amyloid in biological samples: not all Aβ detection methods are created equal
editorial
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disease and is the major
cause of dementia in people aged 65 years and older, affecting approximately 2% of
the population of industrialized countries (Mattson, 2004). The two primary histopathological
lesions found within the AD brain are intracellular accumulations of hyper-phosphorylated
tau protein, in the form of neurofibrillary tangles (NFTs), and extracellular deposits
that are principally comprised of β-amyloid peptide (Aβ) (Adlard and Cummings, 2004).
Aβ has been proposed to be a primary mediator of both the initiation and progression
of disease (Hardy and Selkoe, 2002), with numerous toxic effects attributed to its
abnormal accumulation within the neuropil (Karran et al., 2011). While there remains
debate as to which precise species of Aβ represents the “toxic moiety” (Selkoe, 2008;
Lublin and Gandy, 2010), the assessment of Aβ from bulk tissue remains one of the
most common analyses conducted within the field, particularly as it relates to the
development of therapeutic approaches to this intractable disease. Throughout our
own investigations it became apparent that different commonly used methodologies for
the quantitation of Aβ would often yield markedly different results. In this opinion
piece we discuss this notion and present evidence that highlights the need to exercise
caution when embarking on an analysis of Aβ, or indeed, when interpreting data from
other papers. We also include some of our own data where we analyzed a common set
of tissues using standard analysis techniques that are used in laboratories throughout
the world, including western blot, ELISA and surface-enhanced laser desorption/ionization
(SELDI) time of flight (TOF) mass spectrometry. These analyses are not meant as a
strict side-by-side comparison of raw data generated by different methodologies, but
rather, are designed to emulate different approaches that may be taken by different
laboratories. Thus, whilst the antibodies, extraction buffers and other aspects of
each technique do vary—this is the real-life situation where different approaches
are taken to achieve the same endpoint analysis of Aβ burden. The resulting differences
are striking, and whilst not necessarily unexpected—it is important that the field
acknowledge this phenomenon and have an appreciation for the complexities involved
in the apparently “simple” assessment of Aβ burden.
Methods
The collection, processing, and storage of human brains for research purposes was
conducted by the Victorian Brain Bank Network. Ethics approval was provided by The
University of Melbourne Human Research Ethics Committee, application number 941478X,
titled “Brain Banking for Neuroscience Research.”
Tissue
A cohort of AD (n = 11 M, n = 3 F; 76 ± 2 years), FTD (n = 2 M, n = 3 F; 63 ± 6 years),
DLB (n = 1 M, n = 4 F; 81 ± 5 years) and control (n = 7 M, n = 6 F; 73 ± 2 years)
cases were obtained from the Victorian Brain Bank Network. Gray matter from the frontal
and temporal cortices were sonicated in PBS and centrifuged at 100,000×g (30 min,
4°C). The protein content of the collected supernatant (soluble material) and pellet
(insoluble material) fractions were assessed using the Pierce BCA protein assay kit
according to manufacturers recommendations. The various fractions were subsequently
analyzed using Western blot, ELISA and SELDI.
Aβ assessment methods
For ELISA—Aβ levels were determined using the DELFIA® Double Capture ELISA as previously
described (Ritchie et al., 2003). For western blots and SELDI-TOF mass spectrometry,
Aβ levels were assessed as previously published (Adlard et al., 2008).
Statistical analysis
A one-way analysis of variance with a Tukey's multiple comparison post-hoc test was
utilized to analyse data in Figures 1A–F, while a paired t-test was utilized to analyse
data in Figure 1G (GraphPad Prism 5.0 d).
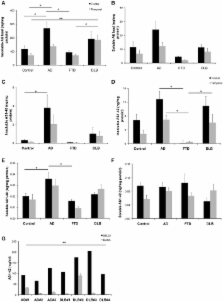
Figure 1
Levels of Aβ detected in brain homogenates from a common set of human cases. (A,B)
Western blot assessment of soluble and insoluble Aβ burden across the entire cohort
of neurological cases and healthy control cases utilized in this study (C–F) ELISA
assessment of soluble and insoluble Aβ burden across the entire cohort of neurological
cases and healthy control cases utilized in this study (G) A comparison of SELDI mass
spectrometric and ELISA assessment of Aβ burden (Aβ 1-40 and Aβ 1-42) across a subset
of the cases examined in this study. *
p < 0.05; **
p < 0.01.
Results
The quantitation of the western blot data for both insoluble and soluble Aβ species
across the different disease states is shown in Figures 1A,B. The highest levels of
both insoluble and soluble Aβ species were present in the AD cases. This is consistent
with the ELISA data for both insoluble and soluble Aβ species across the different
disease states, as shown in Figures 1C–F. A statistical comparison of these data reveal
that the ELISA assay reported significantly (p < 0.0002) lower levels of insoluble
Aβ than the western blot assay (irrespective of whether the ELISA values for Aβ 1-40
and Aβ 1-42 are compared individually or summed for comparison to the western blot
data). Likewise, the soluble data reveal the same difference between assay methodologies
(p = 0.002).
Figure 1G shows a comparison of data generated from a subset of those samples used
above utilizing either SELDI-TOF MS or ELISA techniques for the quantitation of Aβ
across multiple AD and DLB cases. A pairwise comparison of the data generated by the
respective methods for each individual case revealed that SELDI-TOF MS values for
Aβ 1-42 were significantly higher (p < 0.05) than those obtained using ELISA.
Discussion and opinion
In our own studies we had previously noted that common methodologies utilized for
the assessment of protein levels in human biological fluids can return markedly different
results when assessing levels of Aβ peptide. This highlighted the need for careful
methodological considerations when embarking on our own analyses, or when interpreting
the data reported from different laboratories. To formalize this, we assessed a common
set of tissues using different techniques including western blot, ELISA and SELDI-TOF
mass spectrometry, and demonstrated that, under the conditions utilized, both western
blot and SELDI detected significantly larger pools of Aβ in human brain specimens
than did ELISA assays. This was true for both soluble and insoluble fractions of human
brain from a variety of different neurological disease states including AD, FTD, and
DLB cases, as well as from age-matched controls. Whilst the antibodies, extraction
buffers and other aspects of each technique did vary in this comparison—this is the
real-life situation where different approaches are taken in different laboratories
to achieve the same endpoint analysis of Aβ burden. A discussion of these methodological
differences is provided below.
A similar phenomenon has previously been reported when examining tissues from cell
culture media (HEK293 human embryonic kidney cells transfected with a plasmid vector
containing the APP gene with either the Swedish or Arctic and Swedish mutations) and
from transgenic animals (harboring the APP gene with either the Swedish or Arctic
and Swedish mutations), where western blot quantitation revealed a markedly different
Aβ profile than that shown by ELISA assay (Stenh et al., 2005). The authors determined
that ELISA assays are inefficient at measuring Aβ oligomers and that different methodologies
are required for the analysis of soluble Aβ. The data in this study are consistent
with the over-arching finding of this previous report, and clearly demonstrate that
Western blot and SELDI-TOF mass spectrometry are more sensitive techniques that detect
a larger pool of Aβ in the human brain than does ELISA. Furthermore, western blot
and SELDI offer the advantage that more information on the species of Aβ being quantitated
are provided as a function of the methodology, where molecular weight is used to discriminate
the various Aβ species present in a given sample. This represents perhaps one of the
biggest advantages of these techniques over ELISA, which only provides an analysis
of total immunoreactivity to a particular epitope within a sample, with no accounting
for potential cross-reactivity with other proteins. This latter point is an important
caveat, as data generated by ELISA, as with the other methodologies described, is
dependent upon the specific antibodies utilized within the assay. There are a multitude
of different antibodies that have been generated against different epitopes of the
Aβ protein, and these may give different results when used in these assays. Likewise,
as noted above, the specificity of the antibodies to a given target is critical, and
is not always optimal when analysing biological samples. In the case of some commercial
ELISA kits, the precise antibodies utilized (in addition to the extraction buffers
that can liberate different pools of Aβ depending upon their composition) may also
be proprietary, leaving the user essentially blind as to what species of Aβ they are
specifically measuring. This may lead to a situation where the same samples measured
using different ELISA kits provide very different quantitative results. This is a
phenomenon that has been previously reported by Bjerke et al. (2010), who also noted
that the source and quality of the Aβ utilized as a standard within the various ELISA
kits often varies and may provide a source of variance that limits the cross-comparison
of data generated using different ELISA kits. They also demonstrate that there are
numerous confounding factors, other than the use of ELISA, in the analysis of Aβ from
patient populations (the majority which result from the inherent properties of the
Aβ protein itself).
The use of ELISA assays has become common-place in the AD field, representing the
default approach to assay Aβ from a variety of biological fluids. The data presented
in this paper, together with previous anecdotal and literature reports, however, suggest
that the isolated use of ELISA for the quantitation of Aβ may be an insufficient and/or
flawed approach. At a minimum, the reliance on ELISA data may result in an under-representation
of the total Aβ protein levels present in a sample or, in the worse-case scenario,
lead to a misinterpretation of data resulting from clinical trials where the assessment
of efficacy of a given compound is the movement in a particular Aβ species, which
may or may not be within the select “pool” of Aβ detected by an ELISA assay. This
same caveat is also applicable to western blots and SELDI techniques, which are inherently
reliant on discrete antibodies for their methods of detection. The difference, however,
is that these latter techniques generally do not use proprietary antibodies and inherently
provide a greater degree of detail on the precise Aβ species being examined. As the
field continues to move forward, and our understanding of the relative pathogenicity
of different Aβ species crystalizes, it is becoming apparent that the generic “bulk”
assessment of Aβ burden is not sufficiently rigorous to provide the appropriate in-depth
characterization, from both a basic science and a clinical perspective, that is required
in patient populations. The development of new methodologies is critical, and more
precise techniques such as mass spectrometry, which allows for the precise differentiation
and quantitation of relevant analytes in a given sample, are now emerging as the preferred
method for the critical analysis of Aβ from biological samples. Whilst these methods
have their own limitations, in terms of cost, through-put and accessibility to the
necessary infrastructure, their advantages are clear. Until such techniques become
a more readily viable alternative for the field, the discussion and data presented
in this paper highlight the need for a more rigorous approach to the assessment of
Aβ that utilizes more common technologies such as western blot.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial
or financial relationships that could be construed as a potential conflict of interest.
Related collections
Most cited references3
- Record: found
- Abstract: found
- Article: not found
Amyloid-beta oligomers: possible roles as key neurotoxins in Alzheimer's Disease.
Alex Lublin, Sam Gandy (2015)
- Record: found
- Abstract: found
- Article: not found
Amyloid-beta oligomers are inefficiently measured by enzyme-linked immunosorbent assay.
L-G Nilsson, Charlotte Stenh, Pär Gellerfors … (2005)
- Record: found
- Abstract: not found
- Article: not found
Alzheimer's disease--a sum greater than its parts?
Paul A. Adlard, Brian Cummings (2004)