- Record: found
- Abstract: found
- Article: found
Myotonic Dystrophy—A Progeroid Disease?
Read this article at
Abstract
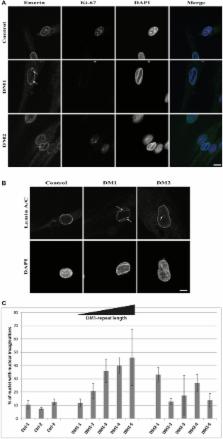
Myotonic dystrophies (DM) are slowly progressing multisystemic disorders caused by repeat expansions in the DMPK or CNBP genes. The multisystemic involvement in DM patients often reflects the appearance of accelerated aging. This is partly due to visible features such as cataracts, muscle weakness, and frontal baldness, but there are also less obvious features like cardiac arrhythmia, diabetes or hypogammaglobulinemia. These aging features suggest the hypothesis that DM could be a segmental progeroid disease. To identify the molecular cause of this characteristic appearance of accelerated aging we compare clinical features of DM to “typical” segmental progeroid disorders caused by mutations in DNA repair or nuclear envelope proteins. Furthermore, we characterize if this premature aging effect is also reflected on the cellular level in DM and investigate overlaps with “classical” progeroid disorders. To investigate the molecular similarities at the cellular level we use primary DM and control cell lines. This analysis reveals many similarities to progeroid syndromes linked to the nuclear envelope. Our comparison on both clinical and molecular levels argues for qualification of DM as a segmental progeroid disorder.
Related collections
Most cited references68
- Record: found
- Abstract: found
- Article: not found
Biological and chemical approaches to diseases of proteostasis deficiency.
- Record: found
- Abstract: found
- Article: not found
Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9.
- Record: found
- Abstract: found
- Article: not found