- Record: found
- Abstract: found
- Article: found
A novel pyroptosis-related lncRNA signature for prognostic prediction in patients with lung adenocarcinoma

Read this article at
ABSTRACT
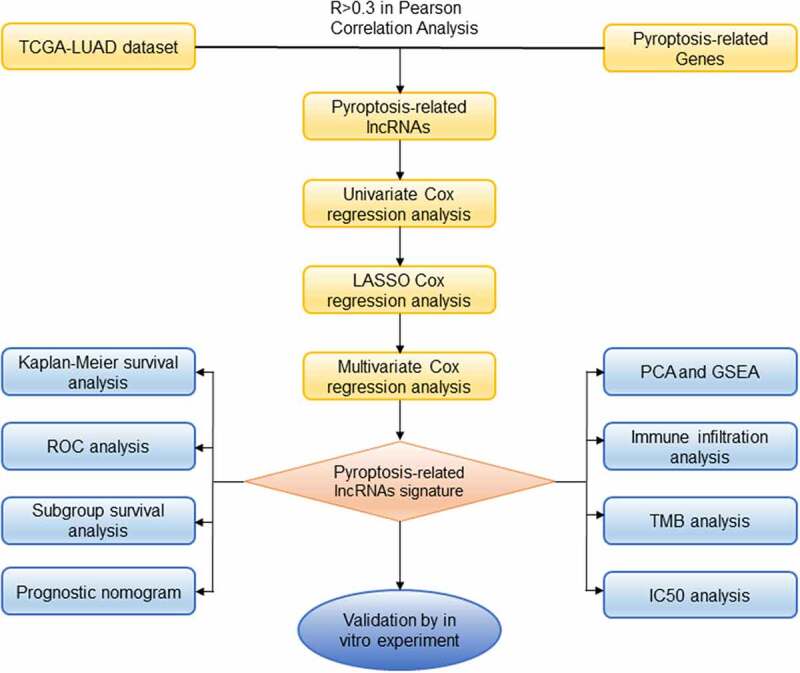
Lung adenocarcinoma (LUAD) has been the major cause of tumor-associated mortality in recent years and has a poor prognosis. Pyroptosis is regulated via the activation of inflammasomes and participates in tumorigenesis. However, the effects of pyroptosis-related lncRNAs (PRlncRNAs) on LUAD have not yet been completely elucidated. Therefore, we attempted to systematically explore patterns of cell pyroptosis to establish a novel signature for predicting LUAD survival. Based on TCGA database, we set up a prognostic model by incorporating PRlncRNAs with differential expression using Cox regression and LASSO regression. Kaplan–Meier analysis was conducted to compare the survival of LUAD patients. We further simplified the risk model and created a nomogram to enhance the prediction of LUAD prognosis. Altogether, 84 PRlncRNAs with differential expression were discovered. Subsequently, a new risk model was constructed based on five PRlncRNAs, GSEC, FAM83A-AS1, AL606489.1, AL034397.3 and AC010980.2. The proposed signature exhibited good performance in prognostic prediction and was related to immunocyte infiltration. The nomogram exactly forecasted the overall survival of patients and had excellent clinical utility. In the present study, the five-lncRNA prognostic risk signature and nomogram are trustworthy and effective indicators for predicting the prognosis of LUAD.
Graphical abstract
Related collections
Most cited references68
- Record: found
- Abstract: found
- Article: not found
Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries
- Record: found
- Abstract: found
- Article: not found
Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles

- Record: found
- Abstract: found
- Article: found