- Record: found
- Abstract: found
- Article: found
A Kalman-Filter Based Approach to Identification of Time-Varying Gene Regulatory Networks
Read this article at
Abstract
Motivation
Conventional identification methods for gene regulatory networks (GRNs) have overwhelmingly adopted static topology models, which remains unchanged over time to represent the underlying molecular interactions of a biological system. However, GRNs are dynamic in response to physiological and environmental changes. Although there is a rich literature in modeling static or temporally invariant networks, how to systematically recover these temporally changing networks remains a major and significant pressing challenge. The purpose of this study is to suggest a two-step strategy that recovers time-varying GRNs.
Results
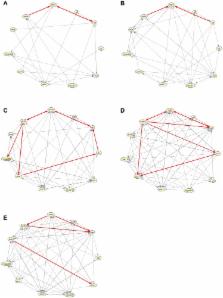
It is suggested in this paper to utilize a switching auto-regressive model to describe the dynamics of time-varying GRNs, and a two-step strategy is proposed to recover the structure of time-varying GRNs. In the first step, the change points are detected by a Kalman-filter based method. The observed time series are divided into several segments using these detection results; and each time series segment belonging to two successive demarcating change points is associated with an individual static regulatory network. In the second step, conditional network structure identification methods are used to reconstruct the topology for each time interval. This two-step strategy efficiently decouples the change point detection problem and the topology inference problem. Simulation results show that the proposed strategy can detect the change points precisely and recover each individual topology structure effectively. Moreover, computation results with the developmental data of Drosophila Melanogaster show that the proposed change point detection procedure is also able to work effectively in real world applications and the change point estimation accuracy exceeds other existing approaches, which means the suggested strategy may also be helpful in solving actual GRN reconstruction problem.
Related collections
Most cited references16
- Record: found
- Abstract: not found
- Article: not found
Genomic analysis of regulatory network dynamics reveals large topological changes.
- Record: found
- Abstract: found
- Article: not found
Gene expression during the life cycle of Drosophila melanogaster.

- Record: found
- Abstract: found
- Article: found