- Record: found
- Abstract: found
- Article: found
Bortezomib antagonizes microtubule-interfering drug-induced apoptosis by inhibiting G2/M transition and MCL-1 degradation
Read this article at
Abstract
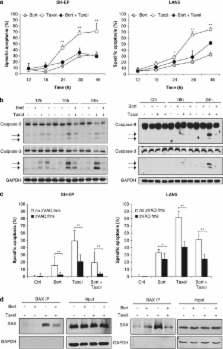
Inhibition of the proteasome is considered as a promising strategy to sensitize cancer cells to apoptosis. Recently, we demonstrated that the proteasome inhibitor Bortezomib primes neuroblastoma cells to TRAIL-induced apoptosis. In the present study, we investigated whether Bortezomib increases chemosensitivity of neuroblastoma cells. Unexpectedly, we discover an antagonistic interaction of Bortezomib and microtubule-interfering drugs. Bortezomib significantly attenuates the loss of cell viability and induction of apoptosis on treatment with Taxol and different vinca alkaloids but not with other chemotherapeutics, that is, Doxorubicin and Cisplatinum. Importantly, Bortezomib inhibits G2/M transition by inhibiting proteasomal degradation of cell cycle regulatory proteins such as p21, thereby preventing cells to enter mitosis, the cell cycle phase in which they are most vulnerable to antitubulin chemotherapeutics. Consequently, Bortezomib counteracts Taxol-induced mitotic arrest and polyploidy, as shown by reduced expression of PLK1 and phosphorylated histone H3. In addition, Bortezomib antagonizes Taxol-mediated degradation of MCL-1 during mitotic arrest by preventing cells to enter mitosis and by inhibiting the proteasome. Downregulation of MCL-1 is critically required for Taxol-induced apoptosis, as overexpression of a phosphomutant MCL-1 variant, which is resistant to degradation, significantly diminishes Taxol-triggered apoptosis. Vice versa, attenuation of Bortezomib-mediated accumulation of MCL-1 by knockdown of MCL-1 significantly enhances Taxol/Bortezomib-induced apoptosis. Thus, Bortezomib rescues Taxol-induced apoptosis by inhibiting G2/M transition and mitigating MCL-1 degradation. The identification of this antagonistic interaction of Bortezomib and microtubule-targeted drugs has important implications for the design of Bortezomib-based combination therapies.
Related collections
Most cited references29
- Record: found
- Abstract: found
- Article: not found
Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy.
- Record: found
- Abstract: found
- Article: not found
Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest.
- Record: found
- Abstract: found
- Article: not found