- Record: found
- Abstract: found
- Article: found
MliR, a novel MerR-like regulator of iron homeostasis, impacts metabolism, membrane remodeling, and cell adhesion in the marine Bacteroidetes Bizionia argentinensis
Read this article at
Abstract
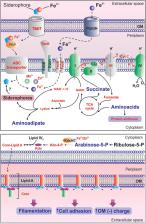
The MerR family is a group of transcriptional activators with conserved N-terminal helix-turn-helix DNA binding domains and variable C-terminal effector binding regions. In most MerR proteins the effector binding domain (EBD) contains a cysteine center suited for metal binding and mediates the response to environmental stimuli, such as oxidative stress, heavy metals or antibiotics. We here present a novel transcriptional regulator classified in the MerR superfamily that lacks an EBD domain and has neither conserved metal binding sites nor cysteine residues. This regulator from the psychrotolerant bacteria Bizionia argentinensis JUB59 is involved in iron homeostasis and was named MliR (MerR-like iron responsive Regulator). In silico analysis revealed that homologs of the MliR protein are widely distributed among different bacterial species. Deletion of the mliR gene led to decreased cell growth, increased cell adhesion and filamentation. Genome-wide transcriptomic analysis showed that genes associated with iron homeostasis were downregulated in mliR-deletion mutant. Through nuclear magnetic resonance-based metabolomics, ICP-MS, fluorescence microscopy and biochemical analysis we evaluated metabolic and phenotypic changes associated with mliR deletion. This work provides the first evidence of a MerR-family regulator involved in iron homeostasis and contributes to expanding our current knowledge on relevant metabolic pathways and cell remodeling mechanisms underlying in the adaptive response to iron availability in bacteria.
Related collections
Most cited references92

- Record: found
- Abstract: found
- Article: found
Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2

- Record: found
- Abstract: found
- Article: found
Trimmomatic: a flexible trimmer for Illumina sequence data

- Record: found
- Abstract: found
- Article: found