- Record: found
- Abstract: found
- Article: found
Astrocyte Unfolded Protein Response Induces a Specific Reactivity State that Causes Non-Cell-Autonomous Neuronal Degeneration
Read this article at
Summary
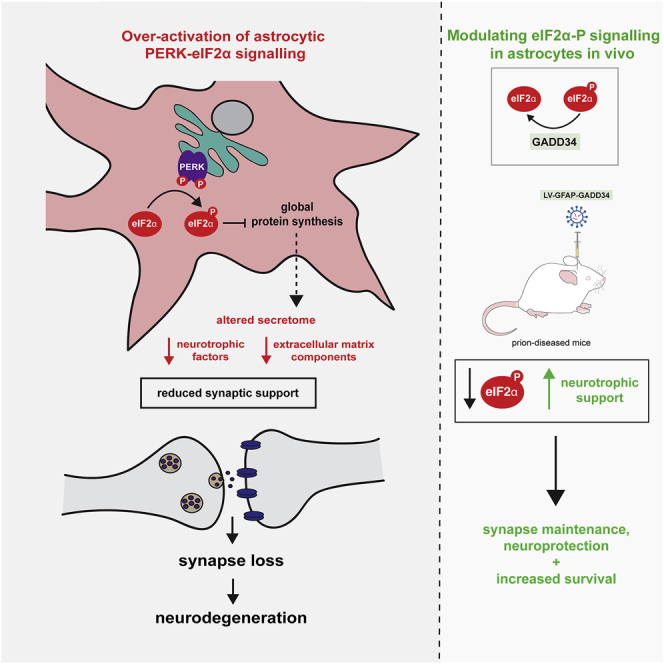
Recent interest in astrocyte activation states has raised the fundamental question of how these cells, normally essential for synapse and neuronal maintenance, become pathogenic. Here, we show that activation of the unfolded protein response (UPR), specifically phosphorylated protein kinase R-like endoplasmic reticulum (ER) kinase (PERK-P) signaling—a pathway that is widely dysregulated in neurodegenerative diseases—generates a distinct reactivity state in astrocytes that alters the astrocytic secretome, leading to loss of synaptogenic function in vitro. Further, we establish that the same PERK-P-dependent astrocyte reactivity state is harmful to neurons in vivo in mice with prion neurodegeneration. Critically, targeting this signaling exclusively in astrocytes during prion disease is alone sufficient to prevent neuronal loss and significantly prolongs survival. Thus, the astrocyte reactivity state resulting from UPR over-activation is a distinct pathogenic mechanism that can by itself be effectively targeted for neuroprotection.
Graphical Abstract
Highlights
-
•
PERK-eIF2α signaling in astrocytes generates a distinct “UPR”-reactivity state
-
•
UPR-reactive astrocytes have an altered secretome, with reduced synaptogenic factors
-
•
UPR-reactive astrocytes fail to support synaptogenesis in vitro
-
•
Targeting the astrocytic UPR prevents synapse and neuronal loss in prion-diseased mice
Abstract
Dysregulation of UPR signaling in neurons is a key mediator of neurodegeneration. Smith et al. show that UPR dysregulation in astrocytes impairs their ability to support synapses; modulating astrocytic UPR signaling prevents neuronal loss and increases survival in prion-diseased mice.
Related collections
Most cited references29
- Record: found
- Abstract: found
- Article: not found
Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis.
- Record: found
- Abstract: found
- Article: not found
Sustained translational repression by eIF2α-P mediates prion neurodegeneration.
- Record: found
- Abstract: found
- Article: not found
Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors
Author and article information
Comments
Comment on this article
See how this article has been cited at scite.ai
scite shows how a scientific paper has been cited by providing the context of the citation, a classification describing whether it supports, mentions, or contrasts the cited claim, and a label indicating in which section the citation was made.