- Record: found
- Abstract: found
- Article: found
The effect of inbreeding, body size and morphology on health in dog breeds
Read this article at
Abstract
Background
Dog breeds are known for their distinctive body shape, size, coat color, head type and behaviors, features that are relatively similar across members of a breed. Unfortunately, dog breeds are also characterized by distinct predispositions to disease. We explored the relationships between inbreeding, morphology and health using genotype based inbreeding estimates, body weight and insurance data for morbidity.
Results
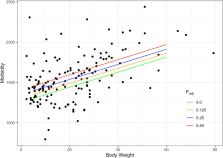
The average inbreeding based on genotype across 227 breeds was F adj = 0.249 (95% CI 0.235–0.263). There were significant differences in morbidity between breeds with low and high inbreeding (H = 16.49, P = 0.0004). There was also a significant difference in morbidity between brachycephalic breeds and non-brachycephalic breeds ( P = 0.0048) and between functionally distinct groups of breeds (H = 14.95 P < 0.0001). Morbidity was modeled using robust regression analysis and both body weight ( P < 0.0001) and inbreeding ( P = 0.013) were significant ( r 2 = 0.77). Smaller less inbred breeds were healthier than larger more inbred breeds.
Related collections
Most cited references59
- Record: found
- Abstract: found
- Article: not found
PLINK: a tool set for whole-genome association and population-based linkage analyses.
- Record: found
- Abstract: found
- Article: not found
A single IGF1 allele is a major determinant of small size in dogs.
- Record: found
- Abstract: found
- Article: not found