- Record: found
- Abstract: found
- Article: not found
von Willebrand factor antigen levels are associated with burden of rare nonsynonymous variants in the VWF gene
Read this article at

Abstract
von Willebrand disease (VWD) is phenotypically heterogeneous, and 35% of patients with type 1 VWD have no known pathogenic von Willebrand factor ( VWF) gene variant. Sadler et al sequenced the entire genomic VWF locus of 737 patients with type 1 VWD or low VWF levels. They report that an accumulation of rare nonsynonymous variants, both pathogenic and nonpathogenic, contributes to the level of VWF and accounts for 31% of the variance in VWF antigen levels.
Key Points
Visual Abstract
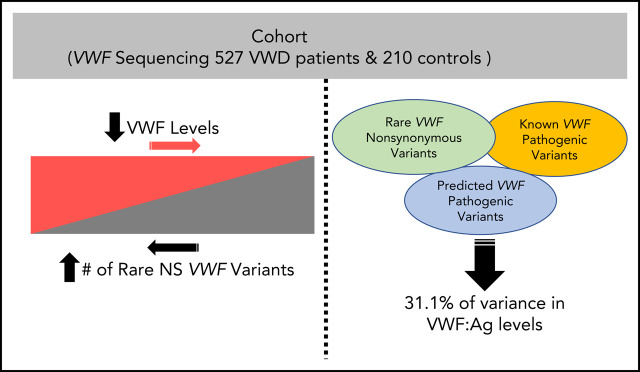
Abstract
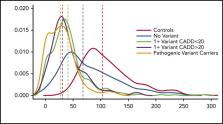
Approximately 35% of patients with type 1 von Willebrand disease (VWD) do not have a known pathogenic variant in the von Willebrand factor ( VWF) gene. We aimed to understand the impact of VWF coding variants on VWD risk and VWF antigen (VWF:Ag) levels, studying 527 patients with low VWF and VWD and 210 healthy controls. VWF sequencing was performed and VWF:Ag levels assayed. A combined annotation-dependent depletion (CADD) score >20 was used as a predicted pathogenicity measure. The number of rare nonsynonymous VWF variants significantly predicted VWF:Ag levels ( P = 1.62 × 10 −21). There was an association between average number of rare nonsynonymous VWF variants with VWD type 1 ( P = 2.4 × 10 −13) and low VWF ( P = 1.6 × 10 −27) compared with healthy subjects: type 1 subjects possessed on average >2 times as many rare variants as those with low VWF and 8 times as many as healthy subjects. The number of rare nonsynonymous variants significantly predicts VWF:Ag levels even after controlling for presence of a variant with a CADD score >20 or a known pathogenic variant in VWF ( P = 2.7 × 10 −14). The number of rare nonsynonymous variants in VWF as well as the presence of a variant with CADD >20 are both significantly associated with VWF levels. The association with rare nonsynonymous variants holds even when controlling for known pathogenic variants, suggesting that additional variants, in VWF or elsewhere, are associated with VWF:Ag levels. Patients with higher VWF:Ag levels with fewer rare nonsynonymous VWF gene variants could benefit from next-generation sequencing to find the cause of their bleeding.
Related collections
Most cited references26

- Record: found
- Abstract: found
- Article: found
CADD: predicting the deleteriousness of variants throughout the human genome
- Record: found
- Abstract: found
- Article: not found
Evolution and functional impact of rare coding variation from deep sequencing of human exomes.
- Record: found
- Abstract: found
- Article: not found