- Record: found
- Abstract: found
- Article: not found
Multifactor Dimensionality Reduction as a Filter-Based Approach for Genome Wide Association Studies
Read this article at

Abstract
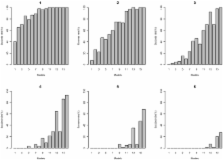
Advances in genotyping technology and the multitude of genetic data available now provide a vast amount of data that is proving to be useful in the quest for a better understanding of human genetic diseases through the study of genetic variation. This has led to the development of approaches such as genome wide association studies (GWAS) designed specifically for interrogating variants across the genome for association with disease, typically by testing single locus, univariate associations. More recently it has been accepted that epistatic (interaction) effects may also be great contributors to these genetic effects, and GWAS methods are now being applied to find epistatic effects. The challenge for these methods still remain in prioritization and interpretation of results, as it has also become standard for initial findings to be independently investigated in replication cohorts or functional studies. This is motivating the development and implementation of filter-based approaches to prioritize variants found to be significant in a discovery stage for follow-up for replication. Such filters must be able to detect both univariate and interactive effects. In the current study we present and evaluate the use of multifactor dimensionality reduction (MDR) as such a filter, with simulated data and a wide range of effect sizes. Additionally, we compare the performance of the MDR filter to a similar filter approach using logistic regression (LR), the more traditional approach used in GWAS analysis, as well as evaporative cooling (EC)-another prominent machine learning filtering method. The results of our simulation study show that MDR is an effective method for such prioritization, and that it can detect main effects, and interactions with or without marginal effects. Importantly, it performed as well as EC and LR for main effect models. It also significantly outperforms LR for various two-locus epistatic models, while it has equivalent results as EC for the epistatic models. The results of this study demonstrate the potential of MDR as a filter to detect gene–gene interactions in GWAS studies.
Related collections
Most cited references43
- Record: found
- Abstract: found
- Article: not found
A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer.
- Record: found
- Abstract: found
- Article: not found
Genome-wide strategies for detecting multiple loci that influence complex diseases.
- Record: found
- Abstract: found
- Article: not found