- Record: found
- Abstract: found
- Article: found
Carbonate-promoted C–H carboxylation of electron-rich heteroarenes†
Read this article at
Abstract
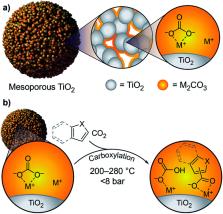
C–H carboxylation is an attractive transformation for both streamlining synthesis and valorizing CO 2. The high bond strength and very low acidity of most C–H bonds, as well as the low reactivity of CO 2, present fundamental challenges for this chemistry. Conventional methods for carboxylation of electron-rich heteroarenes require very strong organic bases to effect C–H deprotonation. Here we show that alkali carbonates (M 2CO 3) dispersed in mesoporous TiO 2 supports (M 2CO 3/TiO 2) effect CO 3 2−-promoted C–H carboxylation of thiophene- and indole-based heteroarenes in gas–solid reactions at 200–320 °C. M 2CO 3/TiO 2 materials are strong bases in this temperature regime, which enables deprotonation of very weakly acidic bonds in these substrates to generate reactive carbanions. In addition, we show that M 2CO 3/TiO 2 enables C3 carboxylation of indole substrates via an apparent electrophilic aromatic substitution mechanism. No carboxylations take place when M 2CO 3/TiO 2 is replaced with un-supported M 2CO 3, demonstrating the critical role of carbonate dispersion and disruption of the M 2CO 3 lattice. After carboxylation, treatment of the support-bound carboxylate products with dimethyl carbonate affords isolable esters and the M 2CO 3/TiO 2 material can be regenerated upon heating under vacuum. Our results provide the basis for a closed cycle for the esterification of heteroarenes with CO 2 and dimethyl carbonate.
Abstract
Carboxylation of heteroarenes with CO 2 is achieved using alkali carbonate dispersed in mesoporous titania as a regenerable reagent.

Related collections
Most cited references46
- Record: found
- Abstract: not found
- Article: not found
On the Interpretation of Deuterium Kinetic Isotope Effects in CH Bond Functionalizations by Transition-Metal Complexes
- Record: found
- Abstract: found
- Article: not found
Pi-nucleophilicity in carbon-carbon bond-forming reactions.
- Record: found
- Abstract: found
- Article: not found