- Record: found
- Abstract: found
- Article: found
Toxoplasma gondii Rhoptry Kinase ROP16 Activates STAT3 and STAT6 Resulting in Cytokine Inhibition and Arginase-1-Dependent Growth Control
Read this article at
Abstract
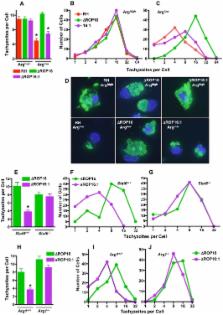
The ROP16 kinase of Toxoplasma gondii is injected into the host cell cytosol where it activates signal transducer and activator of transcription (STAT)-3 and STAT6. Here, we generated a ROP16 deletion mutant on a Type I parasite strain background, as well as a control complementation mutant with restored ROP16 expression. We investigated the biological role of the ROP16 molecule during T. gondii infection. Infection of mouse bone marrow-derived macrophages with rop16-deleted (ΔROP16) parasites resulted in increased amounts of IL-12p40 production relative to the ROP16-positive RH parental strain. High level IL-12p40 production in ΔROP16 infection was dependent on the host cell adaptor molecule MyD88, but surprisingly was independent of any previously recognized T. gondii triggered pathway linking to MyD88 (TLR2, TLR4, TLR9, TLR11, IL-1ß and IL-18). In addition, ROP16 was found to mediate the suppressive effects of Toxoplasma on LPS-induced cytokine synthesis in macrophages and on IFN-γ-induced nitric oxide production by astrocytes and microglial cells. Furthermore, ROP16 triggered synthesis of host cell arginase-1 in a STAT6-dependent manner. In fibroblasts and macrophages, failure to induce arginase-1 by ΔROP16 tachyzoites resulted in resistance to starvation conditions of limiting arginine, an essential amino acid for replication and virulence of this parasite. ΔROP16 tachyzoites that failed to induce host cell arginase-1 displayed increased replication and dissemination during in vivo infection. We conclude that encounter between Toxoplasma ROP16 and the host cell STAT signaling cascade has pleiotropic downstream effects that act in multiple and complex ways to direct the course of infection.
Author Summary
Toxoplasma gondii is an extremely widespread intracellular protozoan parasite that establishes long-lasting infection in humans and animals. Because Toxoplasma infection is most often asymptomatic, it is evident that this parasite has developed sophisticated ways to manipulate host immunity. Recently, the parasite ROP16 kinase was identified as an important determinant of host cell signaling. During cell invasion, ROP16 is injected into the host cell cytoplasm and subsequently localizes to the nucleus. Here, we report the generation of ROP16 knockout parasites (ΔROP16) as well as ΔROP16 complementation mutants (ΔROP16:1) and we describe the biological effects of deleting and re-inserting this molecule. We find that ROP16 controls the ability to activate multiple host cell signaling pathways and simultaneously suppress macrophage proinflammatory responses. Deletion of ROP16 increases parasite ability to replicate and disseminate during in vivo infection. This increased growth response may arise from ROP16-dependent activation of host arginase-1. Induction of arginase-1 limits availability of arginine, an amino acid that is required for parasite growth and host-inducible nitric oxide production. Our results provide new insight into the complex interactions between an intracellular eukaryotic pathogen and its host cell.
Related collections
Most cited references60
- Record: found
- Abstract: found
- Article: not found
The JAK-STAT signaling pathway: input and output integration.
- Record: found
- Abstract: found
- Article: not found
Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens.
- Record: found
- Abstract: found
- Article: not found